science revisited
Mitochondria are essential for the production of adenosine triphosphate (ATP), the energy currency of the cell. Reducing equivalents (electrons), mainly derived from the catabolism of fatty acids and carbohydrates, are shuttled through the four multisubunit complexes (I–IV) of the respiratory chain, embedded in the mitochondrial inner membrane, and generate a proton gradient that is utilized by complex V to generate ATP, a process known as oxidative phosphorylation (OXPHOS).
Mitochondria are the products of mitochondrial DNA (mtDNA) and nuclear DNA (nDNA); mutations in either genome can cause human disease. More than 200 point mutations and hundreds of deletion mutations of mtDNA have been reported. The mitochondrial genome is maternally inherited; therefore, mtDNA defects are typically transmitted from mothers to all progeny. Phenotypic expression of mtDNA defects depends upon heteroplasmy (amount) and tissue distribution of the mutation. The level of heteroplasmy must exceed a critical level (threshold) to produce biochemical and clinical effects. Mutations of mtDNA are designated by “m.” followed by the nucleotide position and change (e.g. m.3243A>G).
As mtDNA maintenance depends on numerous factors encoded by nDNA, there is a variety of defects of intergenomic communication, primary nuclear gene disorders that cause mtDNA instability. The instability of the mitochondrial genome primarily manifests as mtDNA depletion, multiple deletions, or both.
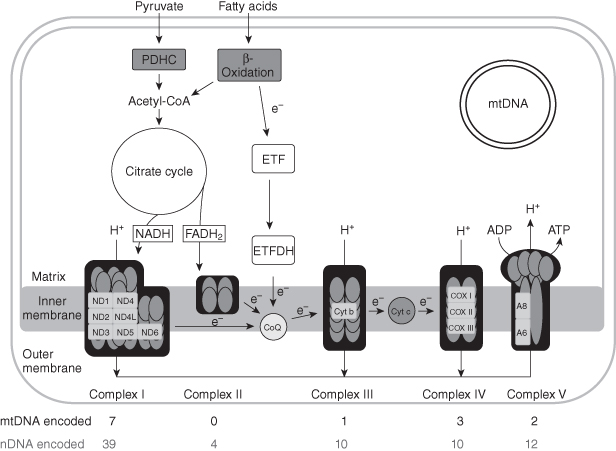
Figure 6.1. Schematic representation of mitochondrial metabolism. Respiratory chain components or complexes encoded by nuclear DNA are grey ovals; subunits encoded by mitochondrial DNA are white rectangles. CoQ, coenzyme Q; Cyt c, cytochrome c; ETF, electron-transferring flavoprotein; ETFDH, electron-transferring flavoprotein dehydrogenase; FADH2, flavin adenine dinucleotide (reduced form); NADH, nicotinamide adenine dinucleotide (reduced form); ND, NADH dehydrogenase; PDHC, pyruvate dehydrogenase complex.
General Clinical Features
Virtually every organ system can be affected by mitochondrial dysfunction and as a consequence, mitochondrial disorders often present as complex multisystemic diseases. Despite their protean clinical presentations, mitochondrial diseases manifest specific clinical symptoms and signs that should alert clinicians to suspect a mitochondrial disease (Box 6.1). Furthermore, particular combinations of clinical manifestations define distinct disorders such as: Kearns–Sayre syndrome (KSS), mitochondrial encephalomyopathy with stroke-like episodes (MELAS), myoclonus epilepsy with ragged-red fibers (MERRF), and mitochondrial neurogastrointestinal encephalomyopathy (MNGIE).
Box 6.1. Clinical features of mitochondrial encephalomyopathies
Muscle
Exercise intolerance
External ophthalmoplegia
Ptosis
Oropharyngeal weakness
Limb weakness
Decreased muscle bulk
Elevated creatine kinasea
Myoglobinuria
Respiratory muscle weakness
Central Nervous System
Leigh syndrome
Epilepsy (partial, generalized, or myoclonus)
Myoclonus
Migraine-like headaches
Stroke-like episodes at a young age
Ataxia
Optic neuropathy
Pigmentary retinopathy
Dementia
Learning disability
Leukodystrophy
Extrapyramidal signs
Motor neuron disease
Peripheral Nervous System
Sensorimotor neuropathy
Endocrine Systems
Diabetes mellitus
Hypothyroidism
Growth hormone deficiency with short stature
Hypoparathyroidism
Delayed puberty
Infertility
Irregular menses
Hirsutism
Heart
Cardiac conduction block
Hypertrophic cardiomyopathy
Pre-excitation syndrome
Renal
Tubular acidosis (de Toni–Fanconi–Debré syndrome)
Bartter-like syndrome
Pancreas
Exocrine deficiency
Liver
Elevated transaminases
Steatosis
Hepatic failure
Hematological
Pancytopenia
Sideroblastic anemia
Gastrointestinal Tract
Dysmotility
Intestinal pseudoobstruction
Psychiatric
Depression
Schizophrenia-like episodes
Dermatological
Purpuric lesions
Hirsutism
Other
Cataracts
Lipomas
aUsually mild except in mtDNA depletion syndrome.
Among the pure or predominantly myopathic forms of mitochondrial diseases, ptosis, progressive external ophthalmoplegia (PEO), or both are particularly common. Extraocular muscles contain abundant quantities of mitochondria and are vulnerable to defects of the mitochondrial respiratory chain. The onset of ptosis and PEO is usually insidious and symmetric, so diplopia and blurred vision are often mild or absent.
Skeletal myopathy is also common. As with most myopathies, neck flexor and proximal limb muscles are disproportionately weaker than other muscles. Serum creatine kinase (CK) levels may be normal or elevated. Many patients complain of premature fatigue out of proportion to the weakness. This exercise intolerance is intuitively compatible with a defect of oxidative phosphorylation (OXPHOS), but challenging to diagnose due to its subjective nature. In such cases, formal exercise testing can be helpful.
Along with muscle, the nervous system is also frequently affected, and consequently, mitochondrial diseases often present as encephalomyopathies. Common central nervous system manifestations include: epilepsy, myoclonus, migraine headaches, stroke-like episodes at a young age, ataxia, optic neuropathy, pigmentary retinopathy, dementia, and psychomotor regression. Peripheral neuropathies are frequent, but often overlooked, in mitochondrial disease patients. The neuropathy is typically axonal, but is mainly demyelinating in MNGIE. Sensorineural hearing loss is commonly associated with mitochondrial encephalomyopathies.
Visceral organ systems affected in mitochondrial diseases include the gastrointestinal system and liver manifesting as gastrointestinal dysmotility and hepatic steatosis. Heart involvement includes cardiomyopathy (often starting as hypertrophic cardiomyopathy), cardiac conduction block, or pre-excitation (Wolff–Parkinson–White) syndrome. Among mitochondrial endocrinopathies, diabetes mellitus is particularly common, but hypothyroidism, growth hormone deficiency, and hypoparathyroidism also occur. Box 6.2 defines the mitochondrial encephalopathies.
Box 6.2. Clinical definitions of mitochondrial encephalomyopathies
Progressive External Ophthalmoplegia (PEO)
Ptosis and PEO generally beginning in childhood or young adulthood
Mitochondrial myopathy (ragged-red and COX-deficient fibers)
Oropharyngeal, facial, and limb myopathy may be present
Kearns–Sayre Syndrome
Onset before age 20
Ophthalmoparesis
Pigmentary retinopathy
Plus, at least one of the following:
CSF protein >100 mg/dL
Cardiac conduction block
Cerebellar syndrome
Sensory Ataxic Neuropathy, Dysarthria, Ophthalmoplegia (SANDO)
Sensory ataxia
Peripheral neuropathy
Dysarthria
PEO
Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS) Syndrome
Stroke at a young age (typically before age 40 years)
Encephalopathy (seizures, dementia, or both)
Ragged-red fibers, lactic acidosis at rest, or both
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


