Clinical Localization of the Lesion
Neuromuscular Transmission Disorders
Spinal Cord Disorders (Myelopathy)
Acute Disseminated Encephalomyelitis
Other Infective or Inflammatory Myelopathies
Chronic Adhesive Arachnoiditis
Spinal Epidural or Subdural Hemorrhage
Arteriovenous Malformation (AVM) or Fistula
Motor Neuron Disease in Children
Motor Neuron Disease in Adults
Other Noninfective Anterior Horn Cell Disorders
Infective Anterior Horn Cell Disorders
Acute Intervertebral Disk Prolapse
Traumatic Avulsion of Nerve Roots
Neuralgic Amyotrophy (Idiopathic Brachial Plexopathy)
Other Causes of Brachial Plexopathy
Acute Inflammatory Polyradiculoneuropathy
(Guillain-Barré Syndrome)
Critical Illness Polyneuropathy
Acute Arsenic or Thallium Poisoning
Organophosphate Polyneuropathy
Disorders of Neuromuscular Transmission
Myasthenic Syndrome (Lambert-Eaton Syndrome)
Emery-Dreifuss Muscular Dystrophy
Trichinosis, Toxoplasmosis, & Sarcoidosis
Polymyositis & Dermatomyositis
Acute Necrotizing Alcoholic Myopathy
Motor-Unit Hyperactivity States
Central Nervous System Disorders
APPROACH TO DIAGNOSIS
Normal motor function depends on the transmission of signals from the brain to the brainstem or spinal cord by upper motor neurons, and from there to skeletal muscle by lower motor neurons (Figure 9-1). A lesion that involves this pathway anywhere along its length may impair motor function. Anatomic structures involved in the regulation or execution of motor activity include the pyramidal and extrapyramidal systems, cerebellum, and lower motor neurons in the cranial nerve nuclei of the brainstem and anterior horns of the spinal cord.
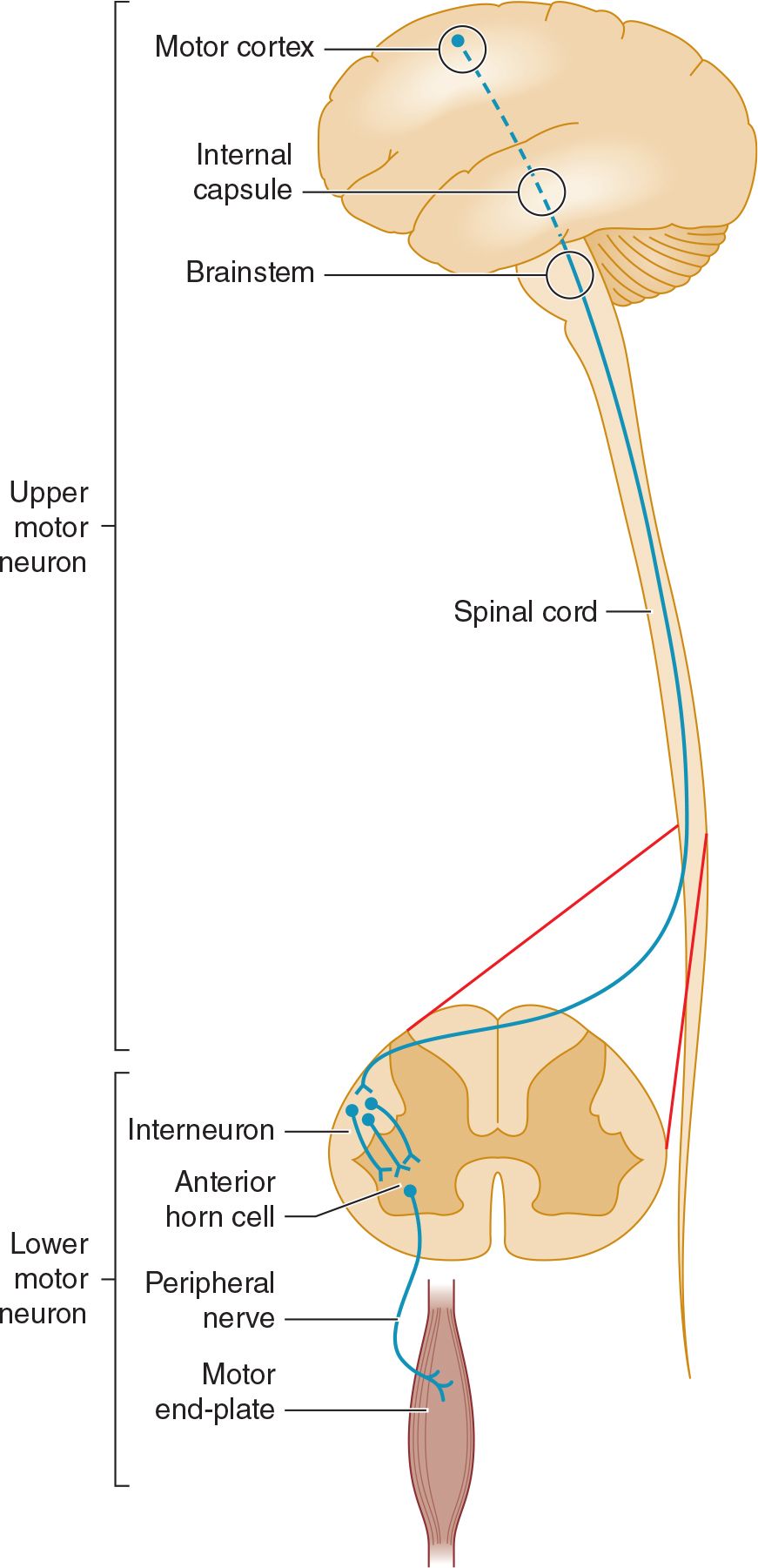
![]() Figure 9-1. Anatomic basis of the upper motor neuron and lower motor neuron concepts.
Figure 9-1. Anatomic basis of the upper motor neuron and lower motor neuron concepts.
The pyramidal system (Figure 9-2) consists of upper motor neuron fibers that descend from the cerebral cortex through the internal capsule, traverse the medullary pyramid, and then mostly decussate, to descend in the lateral corticospinal tract on the opposite side, where they synapse on interneurons and lower motor neurons in the spinal cord.
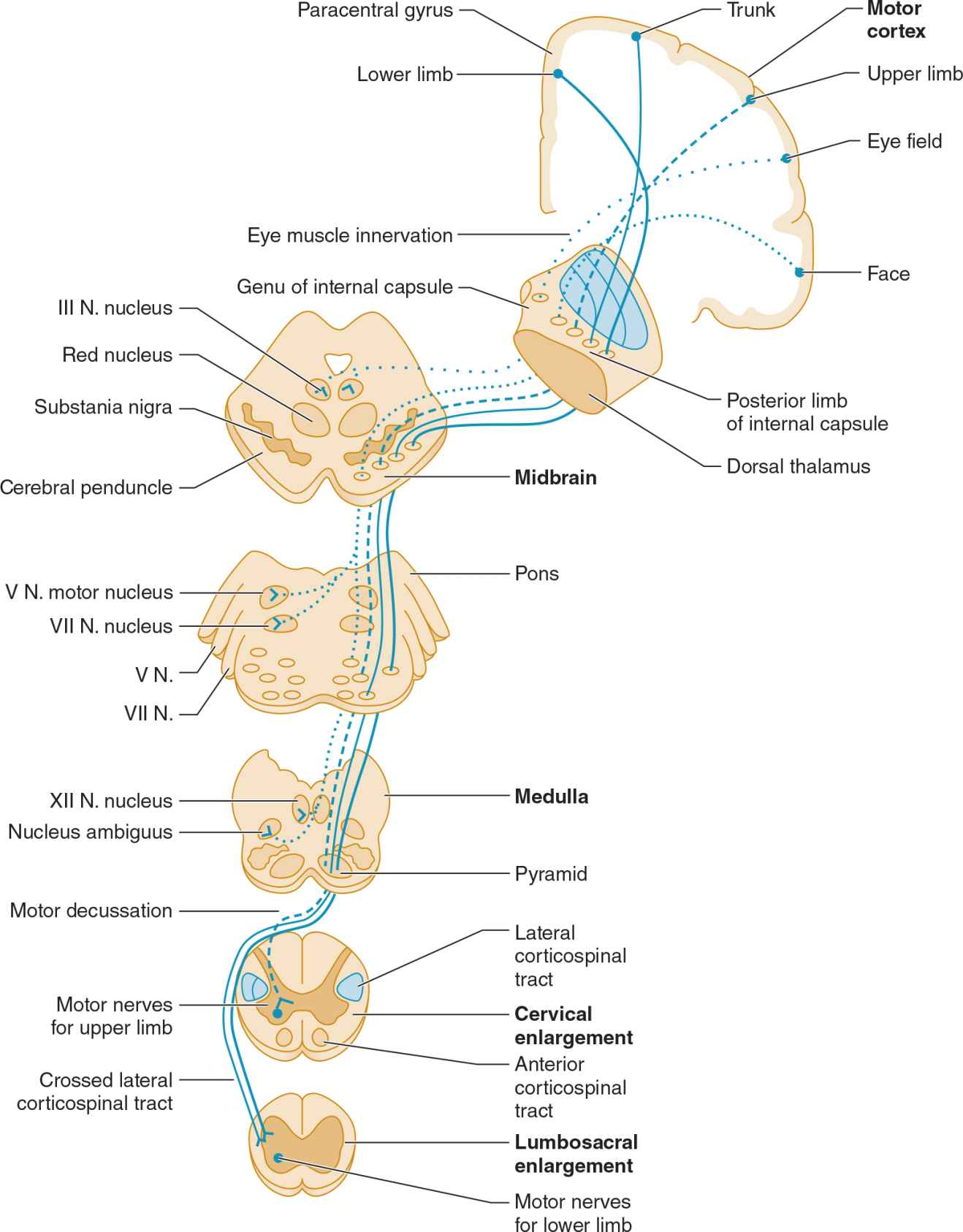
![]() Figure 9-2. Upper motor neuron pathways. Tracts at bottom left are shown outside the cord for clarity only. (Used with permission from McPhee SJ, Hammer GD: Pathophysiology of Disease: An Introduction to Clinical Medicine. 6th ed. New York, NY: McGraw-Hill; 2009.)
Figure 9-2. Upper motor neuron pathways. Tracts at bottom left are shown outside the cord for clarity only. (Used with permission from McPhee SJ, Hammer GD: Pathophysiology of Disease: An Introduction to Clinical Medicine. 6th ed. New York, NY: McGraw-Hill; 2009.)
All other descending influences on lower motor neurons belong to the extrapyramidal system and originate primarily in the basal ganglia and cerebellum. Disorders of the basal ganglia (see Chapter 11, Movement Disorders) and cerebellum (see Chapter 8, Disorders of Equilibrium) are considered separately.
The motor fibers in the cranial and peripheral nerves arise from the lower motor neurons (Figure 9-3). Dysfunction at any point in the peripheral nervous system (anterior horn cell, nerve root, limb plexus, peripheral nerve, or neuromuscular junction) can impair motor function, as can disease of the muscles.
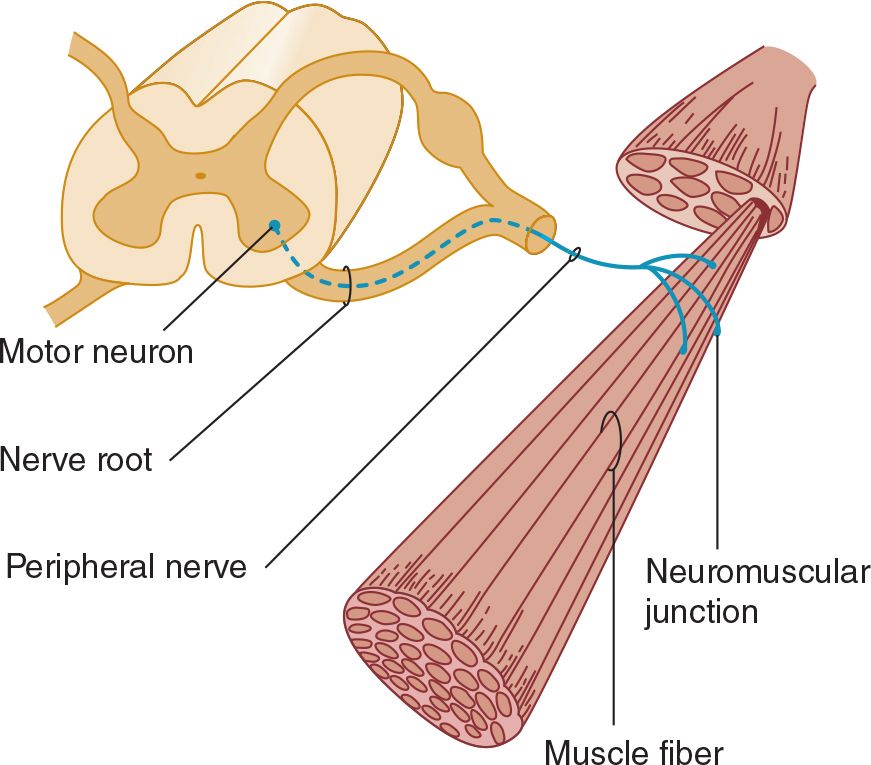
![]() Figure 9-3. Anatomic components of the motor unit.
Figure 9-3. Anatomic components of the motor unit.
HISTORY
Patients with motor deficits generally complain of weakness, heaviness, stiffness, clumsiness, impaired muscular control, or difficulty in executing movements. The term weakness is sometimes used in a nonspecific way to denote fatigue or loss of energy, drive, or enthusiasm, and its meaning must therefore always be clarified. The word is properly used to mean loss of muscle power, and it is in this sense that it is employed here.
HISTORY OF PRESENT ILLNESS
 Mode of Onset
Mode of Onset
An abrupt onset suggests a vascular disturbance, such as a stroke, or certain toxic or metabolic disturbances, whereas subacute onset over days to weeks is commonly associated with a neoplastic, infective, or inflammatory process (Table 9-1). Weakness that evolves slowly over several months or years often has a hereditary, degenerative, endocrinologic, or neoplastic basis.
Table 9-1. Some Causes of Weakness of Acute or Subacute Onset.
Supraspinal lesions
Stroke
Other structural lesions
Spinal cord lesions
Infective: human immunodeficiency virus (HIV) infection
Inflammatory: transverse myelitis, multiple sclerosis
Compressive: tumor, disk protrusion, abscess
Vascular: infarction, hematomyelia
Anterior horn cell disorders
Poliovirus, coxsackievirus, West Nile virus infection
Peripheral nerve disorders
Guillain-Barré syndrome
Diphtheria
Paralytic shellfish poisoning
Porphyria
Arsenic poisoning
Organophosphate toxicity
Neuromuscular junction disorders
Myasthenia gravis
Botulism
Aminoglycoside toxicity
Muscle disorders
Necrotizing myopathies
Acute hypo- or hyperkalemia
Periodic paralyses
 Course
Course
A progressive increase in the motor deficit from its onset suggests continuing activity of the underlying process. Episodic progression suggests a vascular or inflammatory origin; a steadily progressive course is more suggestive of neoplastic disorder or such degenerative conditions as motor neuron disease. Rapid fluctuation of symptoms over short periods (eg, over the course of the day) is characteristic of myasthenia gravis.
 Distribution of Symptoms
Distribution of Symptoms
The distribution of weakness and the presence of associated symptoms may indicate the approximate site of the lesion. For example, weakness in the right arm and leg may result from a lesion of the contralateral motor cortex or the corticospinal pathway at any point above the fifth cervical segment of the spinal cord. Associated right facial weakness indicates that the lesion must be above the level of the facial (VII) nerve nucleus in the brainstem, and an accompanying aphasia (see Chapter 1, Neurologic History & Examination) or visual field defect (see Chapter 7, Neuro-Opthalmic Disorders) localizes it to the cerebral hemisphere.
 Associated Symptoms
Associated Symptoms
The presence and distribution of any sensory abnormalities also help in localizing the lesion. Sensory abnormalities lateralized to the same side as weakness suggest a hemispheric lesion; a cortical lesion is implied by sensory neglect or inattention, agraphesthesia (inability to identify by touch a number written on the skin), astereognosis (inability to identify by touch an object placed in the hand), abarognosis (inability to judge the weight of an object placed in the hand), or impaired two-point discrimination, when peripheral sensory function is intact. Sensory loss below a particular segmental level on the trunk suggests a spinal cord lesion, whereas distal sensory changes in the limbs favor a peripheral nerve lesion. Diseases of the anterior horn cells, neuromuscular junctions, or muscles are not accompanied by altered sensation.
The character of any associated symptoms may suggest the nature of the lesion. Thus progressive leg weakness caused by myelopathy is often preceded or accompanied by pain in the back or legs when the myelopathy is due to a compressive lesion—but not when it has a metabolic or hereditary basis.
 Severity of Symptoms
Severity of Symptoms
The functional severity of a motor deficit is evaluated by determining whether there has been any restriction of daily activities, difficulty in performing previously easy tasks, or reduction in exercise tolerance.
The nature of the functional disturbance depends on the muscles involved. Weakness of proximal muscles in the legs leads to difficulty in climbing or descending stairs or in getting up from a squatting position, whereas weakness in the arms leads to difficulty with such tasks as combing the hair. Distal weakness in the arms may lead to clumsiness, difficulty with such fine motor tasks as doing up buttons or tying shoelaces, and eventually the inability to pick up or grasp objects with the hands, so that even eating becomes difficult or impossible.
Involvement of the muscles supplied by the cranial nerves may lead to diplopia (oculomotor [III], trochlear [IV], or abducens [VI] nerve); difficulty in chewing (trigeminal [V] nerve) or sucking, blowing, or grimacing (facial [VII] nerve); or difficulty in swallowing, with nasal regurgitation and dysarthria (glossopharyngeal [IX], vagus [X], and hypoglossal [XII] nerves).
Weakness of the respiratory muscles leads to tachypnea, the use of accessory muscles of respiration, and anxiety at a stage when arterial blood gases are usually still normal. A vital capacity of less than 1 L in an adult generally calls for ventilatory support, especially if weakness is increasing.
PAST MEDICAL HISTORY
The importance of the past history depends on the patient’s present complaint and the nature of any previous illnesses. For example, in a patient with lung cancer, limb weakness may be due to metastasis or to a remote (nonmetastatic) complication of the cancer. Leg weakness in a diabetic may reflect peripheral nerve, plexus, or multiple root involvement, and hand weakness in a myxedematous patient may be associated with carpal tunnel syndrome.
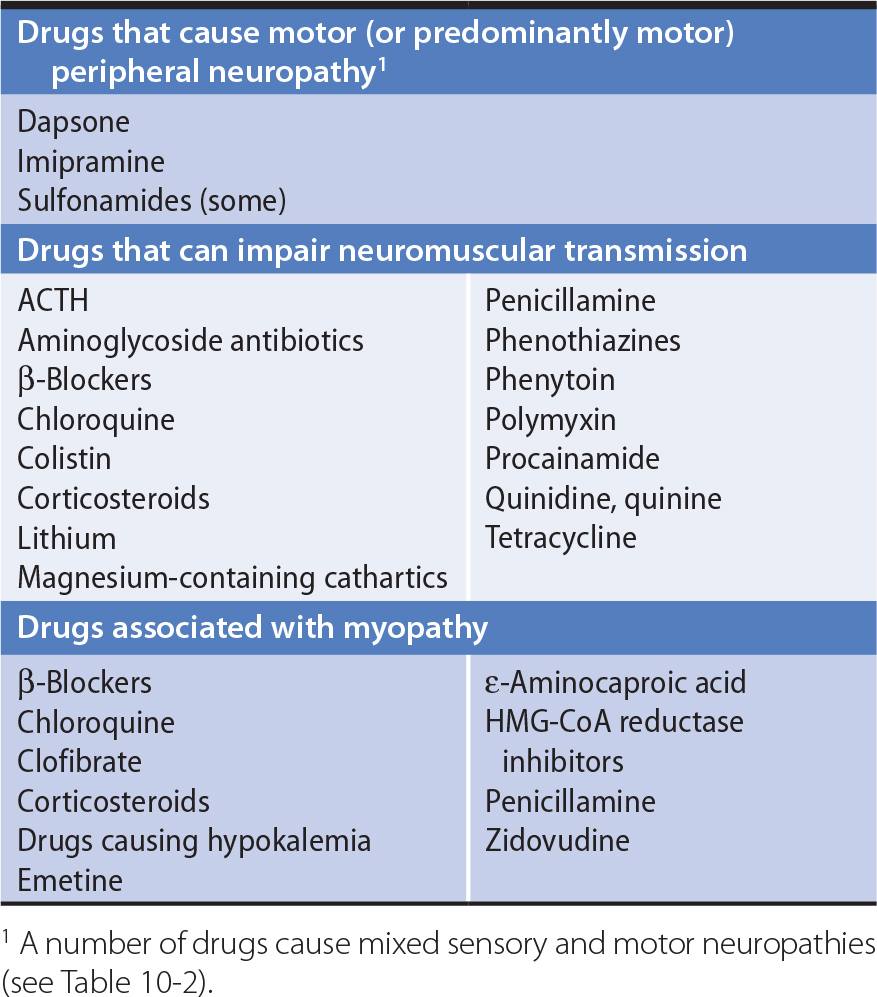
All drugs taken by the patient should be noted. Drugs can cause peripheral neuropathy, impair neuromuscular transmission, or lead to myopathy (Table 9-2).
Table 9-2. Motor Disorders Associated With Drugs.
DEVELOPMENTAL HISTORY
When symptoms develop before adult life, it is particularly important to obtain a full developmental history, including details of the delivery, the birth weight, the patient’s condition in the neonatal period, and the dates at which motor milestones were attained. Congenital or perinatal cerebral disease accounts for most causes of infantile diplegia (weakness of all four limbs, with the legs more severely affected than the arms).
FAMILY HISTORY
Hereditary factors may be important, and the patient’s family background therefore must be explored. Some types of myopathy, motor neuron disease, and peripheral neuropathy have a genetic basis, as do some spinocerebellar degenerations, hereditary spastic paraparesis, and certain other neurologic disorders. It is sometimes necessary to examine other family members to determine whether the patient’s disorder has a hereditary basis.
NEUROLOGIC EXAMINATION
MOTOR SYSTEM
A systematic approach to examining the motor system helps to prevent important abnormalities from being overlooked. A sequential routine for the examination is important.
 Muscle Appearance
Muscle Appearance
1. Wasting, or muscle atrophy, suggests that weakness is due to a lesion of the lower motor neurons or the muscle itself. The distribution of wasting may help to localize the underlying disorder. Upper motor neuron disorders are not usually accompanied by muscle wasting, though atrophy may occasionally occur with prolonged disuse.
2. Pseudohypertrophy of muscles occurs in certain forms of myopathy, but the apparently enlarged muscles are actually weak and flabby.
3. Fasciculations—visible irregular flickerings over the surface of the affected muscle caused by spontaneous contractions of individual motor units—suggest that weakness is due to a lower motor neuron lesion. Fasciculations are most common in anterior horn cell disorders, but may also occur in normal individuals.
4. Flexor or extensor spasms of the limbs are sometimes seen in upper motor neuron disorders as a result of impaired supraspinal control of reflex activity.
 Muscle Tone
Muscle Tone
For clinical purposes, tone can be defined as the resistance of muscle to passive movement of a joint. Tone depends on the degree of muscle contraction and on the mechanical properties of muscle and connective tissue. The degree of muscle contraction depends, in turn, on the activity of anterior horn cells, which is governed by spinal and supraspinal mechanisms.
Tone is assessed by observing the position of the extremities at rest, by palpating the muscle belly, and particularly by determining the resistance to passive stretch and movement. To assess resistance to passive movement, the patient relaxes while each limb is examined in turn by passively taking the major joints through their full range of movement at different speeds and estimating whether the force required is more or less than normal.
Postural abnormalities may result from the increased activity of certain muscle groups caused by disturbances of reflex function, as exemplified by the typical hemiplegic posture—flexion of the upper limb and extension of the ipsilateral lower limb—of many patients who have had a stroke.
1. Hypertonia—Two types of increased tone can be distinguished.
a. Spasticity—consists of an increase in tone that affects different muscle groups to different extents. In the arms, tone is increased more in the flexor and adductor muscles than the extensors and abductors; in the legs, it is greater in the extensor muscles than flexors. The resistance of an affected muscle is not the same throughout the range of movement, but tends to be most marked when passive movement is initiated and then diminishes as the movement continues (the clasp-knife phenomenon). The increase in tone is velocity dependent, so that passive movement at high but not low velocities meets increased resistance. Spasticity is caused by an upper motor neuron lesion, such as a stroke that involves the supplementary motor cortex or corticospinal tract. It may not become apparent for several days after onset of an acute lesion.
b. Rigidity—consists of increased resistance to passive movement that is independent of the direction of the movement; that is, it affects agonist and antagonist muscle groups equally (lead-pipe rigidity). The term cogwheel rigidity is used when there are superimposed ratchet-like interruptions in the passive movement, probably related to underlying tremor. In general, rigidity indicates extrapyramidal dysfunction and is due to a lesion of the basal ganglia (eg, Parkinson disease).
2. Hypotonia (flaccidity)—This is characterized by excessive floppiness—a reduced resistance to passive movement—so that the distal portion of the limb is easily waved to and fro when the extremity is passively shaken. In hypotonic limbs it is often possible to hyperextend the joints, and the muscle belly may look flattened and feel less firm than usual. Although hypotonia usually relates to pathologic involvement of the lower motor neuron supply to the affected muscles, it can also occur with primary muscle disorders, disruption of the sensory (afferent) limb of the reflex arc, cerebellar disease, and certain extrapyramidal disorders such as Huntington disease, as well as in the acute stage of a pyramidal lesion.
3. Paratonia—Some patients seem unable to relax and will move the limb being examined as the physician moves it, despite instructions to the contrary. In more advanced cases, there seems to be rigidity when the examiner moves the limb rapidly but normal tone when the limb is moved slowly. This phenomenon—paratonia—is particularly apt to occur in patients with frontal lobe or diffuse cerebral disease.
 Muscle Power
Muscle Power
To test muscle power, the patient is asked to resist pressure exerted by the examiner. Based on the history and other findings, muscles particularly likely to be affected are selected for initial evaluation, and other muscles are subsequently examined to determine the distribution of weakness more fully and to shorten the list of diagnostic possibilities. For instance, if an upper motor neuron (pyramidal) lesion is suspected, the extensors and abductors of the upper extremity and flexors of the lower extremity are tested in the most detail, because they will be the most affected. Strength on the two sides is compared so that minor degrees of weakness can be recognized.
1. Distinction between upper and lower motor neuron lesions—The distribution of weakness helps to distinguish between dysfunction of the upper or lower motor neurons. Upper motor neuron lesions (eg, stroke) lead to weakness that characteristically involves the extensors and abductors more than the flexors and adductors of the arms—and the flexors more than the extensors of the legs. Lower motor neuron lesions produce weakness of the muscles supplied by the affected neurons; the particular distribution of the weakness may point to lower motor neuron disturbance involving the spinal cord, nerve roots, plexus, or peripheral nerves.
2. Distinction between myopathic and neuropathic disorders—Weakness may also result from a primary muscle disorder (myopathy) or from a disorder of lower motor neurons. In patients with a motor deficit in all limbs that is not due to an upper motor neuron lesion, proximal distribution of weakness suggests a myopathic disorder, whereas predominantly distal involvement suggests a neuropathic disturbance.
3. Neuromuscular junction disorders—Marked variability in the severity and distribution of weakness over short periods of time (eg, over the course of a day) suggests myasthenia gravis, a disorder of neuromuscular transmission.
4. Psychogenic disorders—Apparent weakness that is not organic in nature also shows a characteristic variability; it is often more severe on formal testing than is consistent with the patient’s daily activities. Moreover, palpation of antagonist muscles commonly reveals that they contract each time the patient is asked to activate the agonist.
For practical and comparative purposes, power is best graded in the manner shown in Table 9-3.
Table 9-3. Grading of Muscle Power According to the System Suggested by the Medical Research Council.
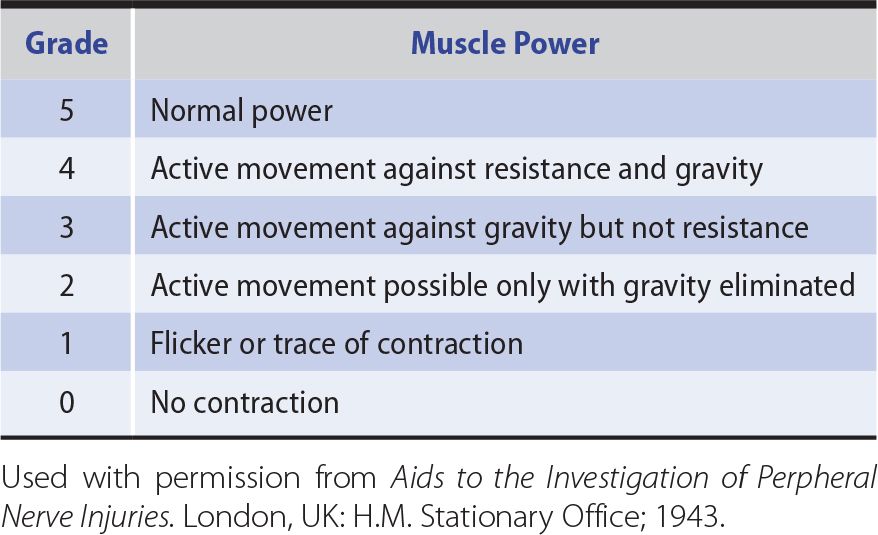
The term monoplegia denotes paralysis or severe weakness of the muscles in one limb, and monoparesis denotes less severe weakness in one limb, although the two words are often used interchangeably. Hemiplegia or hemiparesis is weakness in both limbs (and sometimes the face) on one side of the body; paraplegia or paraparesis is weakness of both legs; and quadriplegia or quadriparesis (also tetraplegia, tetraparesis) is weakness of all four limbs.
COORDINATION
The coordination of motor activity can be impaired by weakness, sensory disturbances, or cerebellar disease and requires careful evaluation.
Voluntary activity is observed with regard to its accuracy, velocity, range, and regularity, and the manner in which individual actions are integrated to produce a smooth complex movement.
In the finger-nose test, the patient moves the index finger to touch the tip of his or her nose and then the tip of the examiner’s index finger; the examiner can move the target finger about during the test to change its location and should position it so that the patient’s arm must extend fully to reach it.
In the heel-knee-shin test, the recumbent patient lifts one leg off the bed, flexes it at the knee, places the heel on the other knee, and runs the heel down the shin as smoothly as possible.
Rapid alternating movement is tested by asking the patient to tap repetitively with one hand on the back of the other, to tap alternately with the palm and back of one hand on the back of the other hand or on the knee, to screw an imaginary light bulb into the ceiling with each arm in turn, and to rub the fingers of one hand in a circular polishing movement on the back of the other hand. Other tests of rapid alternating movement include tapping on the ball of the thumb with the tip of the index finger or tapping the floor as rapidly as possible with the sole while keeping the heel of the foot in place. During all these tests, the examiner looks for irregularities of rate, amplitude, and rhythm and for precision of movements. With pyramidal lesions, fine voluntary movements are performed slowly. With cerebellar lesions, the rate, rhythm, and amplitude of such movements are irregular.
If loss of sensation may be responsible for impaired coordination, the maneuver should be repeated both with eyes closed and with visual attention directed to the limb; with visual feedback the apparent weakness or incoordination will improve. In patients with cerebellar disease, the main complaint and physical finding are often of incoordination, and examination may reveal little else. Further discussion of cerebellar ataxia and the various terms used to describe aspects of it can be found in Chapter 8, Disorders of Equilibrium.
TENDON REFLEXES
Changes in the tendon (muscle stretch) reflexes may accompany disturbances in motor or sensory function and provide a guide to the cause of any motor deficit. The tendon is tapped with a reflex hammer to produce a sudden brisk stretch of the muscle and its contained spindles. The clinically important stretch reflexes and the nerves, roots, and spinal segments subserving them are indicated in Table 9-4. When the reflexes are tested, the limbs on each side should be placed in identical positions and the reflexes elicited in the same manner.
Table 9-4. Tendon (Muscle Stretch) Reflexes.1
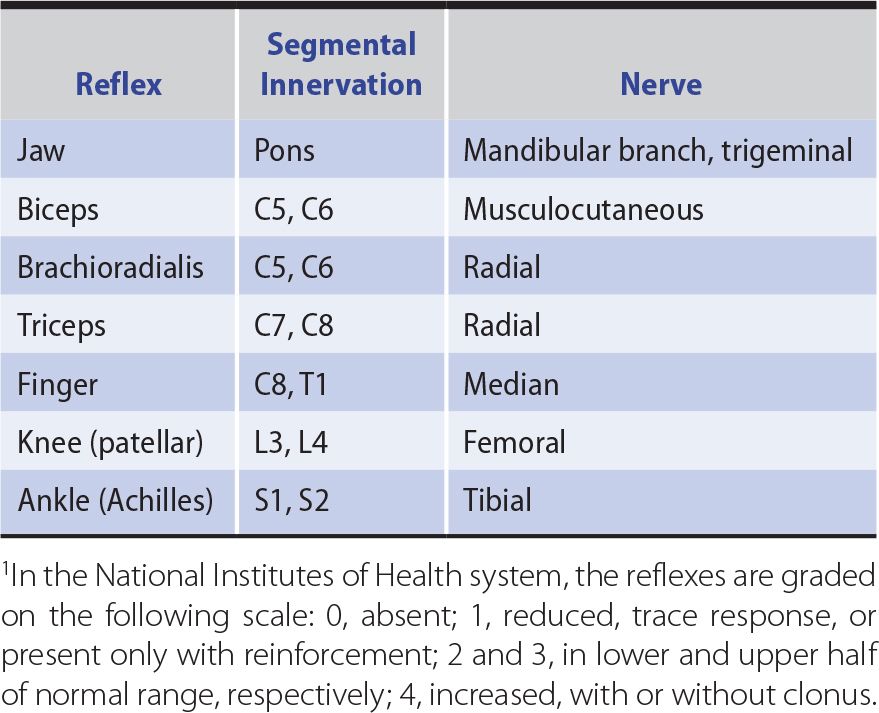
1. Areflexia—Apparent loss of the tendon reflexes may merely reflect a lack of clinical expertise by the examiner. Performance of the Jendrassik maneuver (an attempt by the patient to pull apart the fingers of the two hands when they are hooked together) or some similar distracting action (such as making a fist with the hand that is not being tested) may allow an otherwise unobtainable reflex response to be elicited. A reflex may be lost or depressed by any lesion that interrupts the structural or functional continuity of its reflex arc, as in a root lesion or peripheral neuropathy. In addition, reflexes are often depressed during the acute stage of an upper motor neuron lesion, during deep coma, and with cerebellar disease.
2. Hyperreflexia—Increased reflexes occur with upper motor neuron lesions, but they may also occur with a symmetric distribution in certain healthy subjects and in patients under emotional stress. The presence of reflex asymmetry is therefore of particular clinical significance. Clonus consists of a series of rhythmic reflex contractions of a muscle that is suddenly subjected to sustained stretch, with each beat caused by renewed stretch of the muscle during relaxation from its previous contracted state. Sustained clonus—more than three or four beats in response to sudden sustained stretch—is always pathologic and is associated with an abnormally brisk reflex. In hyperreflexic states, the region from which a particular reflex response can be elicited may be enlarged. For example, elicitation of the biceps reflex may be accompanied by reflex finger flexion, or eliciting the finger flexion reflex may cause flexion of the thumb (Hoffmann sign).
3. Reflex asymmetry—Although the intensity of reflex responses varies considerably among subjects, reflexes should be symmetric in any individual.
a. Lateralized asymmetries of response—reflexes that are brisker on one side of the body than on the other—usually indicate an upper motor neuron disturbance, but sometimes reflect a lower motor neuron lesion on the side with the depressed reflexes.
b. Focal reflex deficits often relate to root, plexus, or peripheral nerve lesions. For example, unilateral depression of the ankle jerk commonly reflects an S1 radiculopathy resulting from a lumbosacral disk lesion.
c. Loss of distal tendon reflexes (especially ankle jerks), with preservation of more proximal ones, is common in polyneuropathies.
SUPERFICIAL REFLEXES
1. The polysynaptic superficial abdominal reflexes, which depend on the integrity of the T8-12 spinal cord segments, are elicited by gently stroking each quadrant of the abdominal wall with a blunt object such as a wooden stick. A normal response consists of contraction of the muscle in the quadrant stimulated, with a brief movement of the umbilicus toward the stimulus. Asymmetric loss of the response may be of diagnostic significance. The response may be depressed or lost on one side in patients with an upper motor neuron disturbance affecting that side. Segmental loss of the response may relate to local disease of the abdominal wall or its innervation, as in a radiculopathy. Bilaterally absent responses are usually of no significance, occurring in the elderly, the obese, multiparous women, and patients who have had abdominal surgery.
2. The cremasteric reflex, mediated through the L1 and L2 reflex arcs, consists of retraction of the ipsilateral testis when the inner aspect of the thigh is lightly stroked; it is lost in patients with a lesion involving these nerve roots. It is also lost in patients with contralateral upper motor neuron disturbances.
3. Stimulation of the lateral border of the foot in a normal adult leads to plantar flexion of the toes and dorsiflexion of the ankle. The Babinski response consists of dorsiflexion of the big toe and fanning of the other toes in response to stroking firmly the lateral border of the foot, which is part of the S1 dermatome; flexion at the hip and knee may also occur. Such an extensor plantar response indicates an upper motor neuron lesion involving the contralateral motor cortex or the corticospinal tract. It can also be found bilaterally in anesthetized or comatose subjects, in patients who have just had a seizure, and in normal infants.
An extensor plantar response can also be elicited, though less reliably, by such maneuvers as pricking the dorsal surface of the big toe with a pin (Bing sign), firmly stroking down the anterior border of the tibia from knee to ankle (Oppenheim maneuver), squeezing the calf muscle (Gordon maneuver), or Achilles tendon (Schafer maneuver), flicking the little toe (Gonda maneuver), or stroking the back of the foot just below the lateral malleolus (Chaddock maneuver). In interpreting responses to these maneuvers, attention must be focused only on the direction in which the big toe first moves.
GAIT
The patient is observed while walking at a comfortable pace. Attention is directed at the stance and posture; the facility with which the patient starts and stops walking and turns to either side; the length of the stride; the rhythm of walking; the presence of normally associated movements, such as swinging of the arms; and any involuntary movements (see Figure 1-25).
Subtle gait disorders become apparent only when the patient is asked to run, walk on the balls of the feet or the heels, hop on either foot, or walk heel-to-toe along a straight line. Gait disorders occur in many neurologic disturbances. A motor or sensory disturbance may lead to an abnormal gait whose nature depends on the site of pathologic involvement.
1. Apraxic gait—Apraxic gait occurs in some patients with disturbances, usually bilateral, of frontal lobe function, such as in hydrocephalus or progressive dementing disorders. There is no weakness or incoordination of the limbs, but the patient is unable to stand unsupported or to walk properly—the feet seem glued to the ground. If walking is possible at all, the gait is unsteady, uncertain, and short-stepped, with marked hesitation (“freezing”), and the legs are moved in a direction inappropriate to the center of gravity.
2. Corticospinal lesions—A corticospinal lesion, irrespective of cause, can cause a gait disturbance.
a. In patients with hemiparesis, the affected leg must be circumducted to be advanced. The patient tilts at the waist toward the normal side and swings the affected leg outward as well as forward, thus compensating for any tendency to drag or catch the foot on the ground because of weakness in the hip and knee flexors or ankle dorsiflexors. The arm on the affected side is usually held flexed and adducted. In mild cases, there may be no more than a tendency to drag the affected leg, so that the sole of that shoe tends to be excessively worn.
b. With severe bilateral spasticity, the legs are brought stiffly forward and adducted, often with compensatory truncal movements (“scissors-like” gait). This gait is seen in its most extreme form in children with spastic diplegia from perinatally acquired static encephalopathy. In patients with mild spastic paraparesis, the gait is shuffling, slow, stiff, and awkward, with the feet tending to drag.
3. Frontal disorders—Some patients with frontal lobe or white matter lesions have a gait characterized by short, shuffling steps; hesitation in starting (“ignition failure”) or turning; unsteadiness; and a wide or narrow base. Sometimes referred to as marche à petit pas, this abnormality may be mistaken for a parkinsonian gait, but the wide base, preserved arm swing, absence of other signs of parkinsonism, and accompanying findings of cognitive impairment, frontal release signs, pseudobulbar palsy, pyramidal deficits, and sphincter disturbances should suggest the correct diagnosis. In patients with frontotemporal dementia, however, a parkinsonian gait and other extrapyramidal findings may be present.
4. Extrapyramidal disorders.
a. Patients with advanced parkinsonism are often stooped, have difficulty in beginning to walk, and may need to lean farther and farther forward while walking in place in order to advance; once in motion, there may be unsteadiness in turning and difficulty in stopping. The gait itself is characterized by small strides, often taken at an increasing rate until the patient is almost running (festination), and by loss of the arm swinging that normally accompanies locomotion. At times, as when walking through a doorway, the patient may be unable to advance (“freezing”). Turning may require several small steps. In mild parkinsonism, a mildly slowed or unsteady gait, flexed posture, or reduced arm swinging may be the only abnormality found.
b. Abnormal posturing of the limbs or trunk is a feature of dystonia; it can interfere with locomotion or lead to a distorted and bizarre gait.
c. Chorea can cause an irregular, unpredictable, and unsteady gait, as the patient dips or lurches from side to side. Choreiform movements of the face and extremities are usually evident.
d. Tremor that occurs primarily on standing (orthostatic tremor) may lead to an unsteady, uncertain gait, with hesitancy in commencing to walk.
5. Cerebellar disorders—Several gait disorders may occur in cerebellar disorders (see Chapter 8, Disorders of Equilibrium).
a. Truncal ataxia results from involvement of midline cerebellar structures, especially the vermis. The gait is irregular, clumsy, unsteady, uncertain, and broad-based, with the patient walking with feet wide apart for additional support. Turning and heel-to-toe walking are especially difficult. There are often few accompanying signs of a cerebellar disturbance in the limbs. Causes include midline cerebellar tumors and the cerebellar degeneration that can occur with alcoholism or hypothyroidism, as a nonmetastatic complication of cancer, and with certain hereditary disorders.
b. In extreme cases, with gross involvement of midline cerebellar structures (especially the vermis), the patient cannot stand without falling.
c. A lesion of one cerebellar hemisphere leads to an unsteady gait in which the patient consistently falls or lurches toward the affected side.
6. Vestibular disorders—With unilateral vestibular dysfunction, the patient is unsteady, veering to the affected side. If both sides are affected, the gait becomes especially unsteady in the dark, when visual input is reduced.
7. Impaired sensation—Impaired sensation, especially proprioception, also leads to an unsteady gait that is aggravated by walking in the dark or with the eyes closed, as visual input cannot then compensate for the sensory loss. Because of their defective position sense, many patients lift their feet higher than necessary when walking, producing a steppage gait. Causes include tabes dorsalis, sensory neuropathies, vitamin B12 deficiency, and certain hereditary disorders (see Chapter 10, Sensory Disorders).
8. Anterior horn cell, peripheral motor nerve, or skeletal muscle disorders—These disorders lead to gait disturbances if the muscles involved in locomotion are affected. Weakness of the anterior tibial muscles leads to foot drop; to avoid catching or scuffing the foot on the ground, the patient must lift the affected leg higher than the other, in a steppage gait resembling that of sensory disorders. Weakness of the calf muscles leads to an inability to walk on the balls of the feet. Weakness of the trunk and girdle muscles, such as occurs in muscular dystrophy, other myopathic disorders, and Kugelberg-Welander syndrome, leads to a waddling gait because the pelvis tends to slump toward the non–weight-bearing side.
9. Unsteady or cautious gait in the elderly—Many elderly persons complain of unsteadiness when walking and of a fear of falling, but neurologic examination reveals no abnormality. Their gait is cautious, unsteady, and sometimes difficult to initiate. Frontal lobe dysfunction may be responsible, as may reduce sensory input from several different afferent systems and impair central processing of sensory input; impaired vestibular function may also be important.
10. Psychogenic gait disorder—This is suggested by fluctuations of stance and gait, commonly in response to suggestion, frequently accompanied by a sudden buckling at the knees, often without falls. Apart from the bizarre gait, neurologic examination may be normal. Patients apparently unable to stand or walk (astasia-abasia) may actually be able to walk on a narrow base, but sway wildly in all directions, often with waving of the arms as if about to fall.
CLINICAL LOCALIZATION OF THE LESION
The findings on examination should indicate whether the motor deficit is due to an upper or lower motor neuron disturbance, impaired neuromuscular transmission, or a primary muscle disorder. With an upper or lower motor neuron disturbance, the clinical findings may also help to localize the lesion to a single level of the nervous system, thereby reducing the number of diagnostic possibilities.
UPPER MOTOR NEURON LESIONS
 Signs
Signs
The following classic signs of an upper motor neuron lesion occur with involvement of the upper motor neuron at any point; further clinical findings depend on the actual site of the lesion.
1. Weakness or paralysis
2. Spasticity
3. Increased tendon reflexes
4. Extensor plantar (Babinski) response
5. Loss of superficial abdominal reflexes
6. Little, if any, muscle atrophy
 Localization of Underlying Lesion
Localization of Underlying Lesion
1. A parasagittal intracranial lesion produces an upper motor neuron deficit that characteristically affects both legs and may later involve the arms.
2. A discrete lesion of the cerebral cortex or its projections may produce a focal motor deficit involving, for example, the contralateral hand. Weakness may be restricted to the contralateral leg in patients with anterior cerebral artery occlusion or to the contralateral face and arm if the middle cerebral artery is involved. A more extensive cortical or subcortical lesion will produce weakness or paralysis of the contralateral face, arm, and leg and may be accompanied by aphasia, a visual field defect, or a sensory disturbance of cortical type.
3. A lesion in the internal capsule, where the descending fibers from the cerebral cortex are closely packed, commonly results in a severe hemiparesis involving the contralateral limbs and face.
4. A brainstem lesion commonly—but not invariably—leads to bilateral motor deficits, often with accompanying sensory and cranial nerve disturbances, and disequilibrium. A more limited brainstem lesion characteristically leads to an ipsilateral cranial nerve disturbance and contralateral hemiparesis; the cranial nerves affected depend on the level at which the brainstem is involved.
5. A unilateral spinal cord lesion above the fifth cervical segment (C5) causes an ipsilateral hemiparesis that spares the face and cranial nerves. Lesions between C5 and the first thoracic segment (T1) affect the ipsilateral arm to a variable extent as well as the ipsilateral leg; a lesion below T1 affects only the ipsilateral leg. If both sides of the spinal cord are involved, quadriparesis or paraparesis usually results. Increased muscle tone (spasticity) may be more prominent than weakness. If there is an extensive but unilateral cord lesion, the motor deficit is accompanied by ipsilateral impairment of vibration and position sense and by contralateral loss of pain and temperature appreciation (Brown-Séquard syndrome).
6. With compressive and other focal lesions that involve the anterior horn cells in addition to the fiber tracts traversing the cord, the muscles innervated by the affected cord segment weaken and atrophy. Therefore, a focal lower motor neuron deficit exists at the level of the lesion, and an upper motor neuron deficit exists below it—in addition to any associated sensory disturbance.
LOWER MOTOR NEURON LESIONS
 Signs
Signs
Lower motor neuron lesions produce the following characteristic signs at the affected levels.
1. Weakness or paralysis
2. Wasting and fasciculations of involved muscles
3. Hypotonia (flaccidity)
4. Loss of tendon reflexes when neurons subserving them are affected
5. Normal abdominal and plantar reflexes—unless the neurons subserving them are directly involved, in which case reflex responses are lost
 Localization of the Underlying Lesion
Localization of the Underlying Lesion
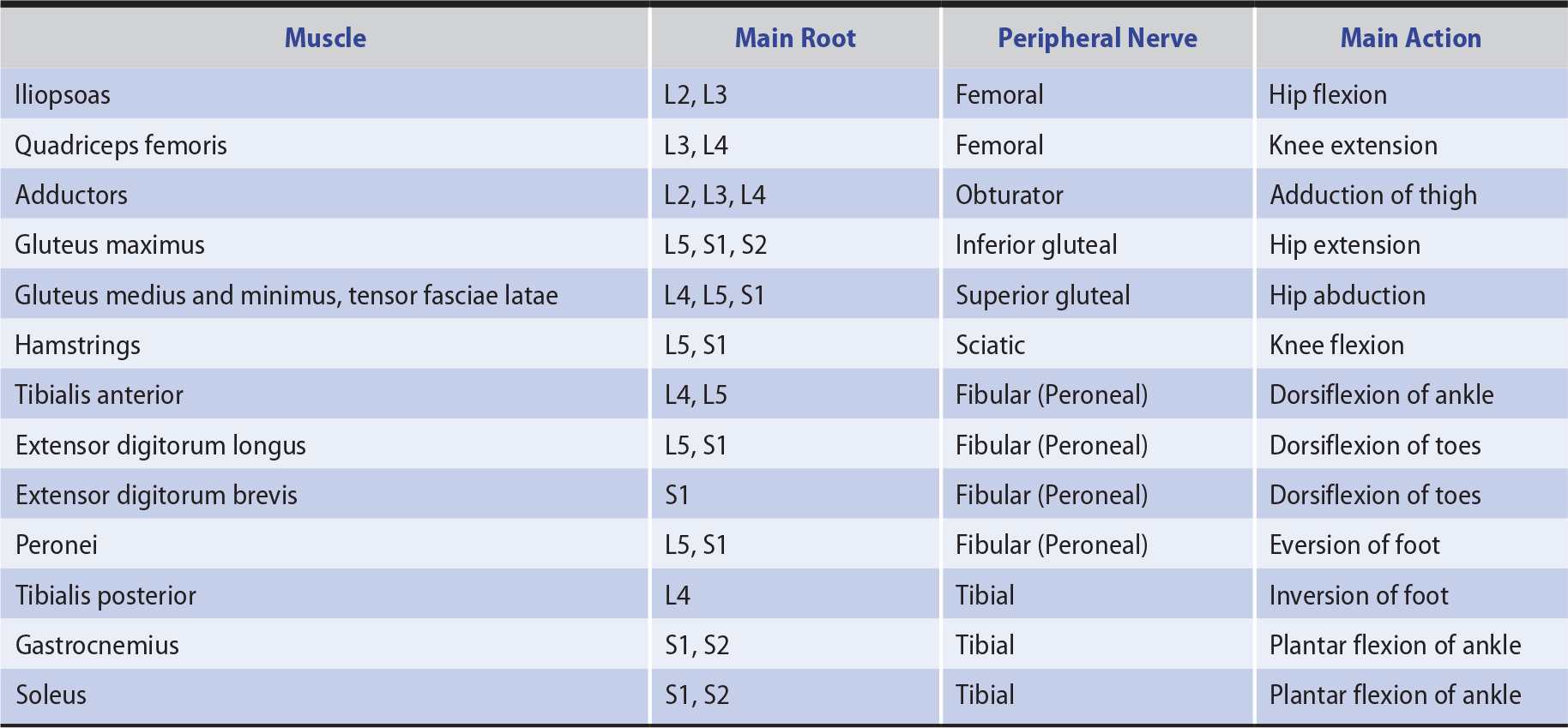
In distinguishing weakness from a segmental spinal cord, anterior horn cell, radicular (nerve root), plexus, or peripheral nerve lesion, the distribution of the motor deficit is important. Only those muscles supplied wholly or partly by the involved structure are weak (Tables 9-5 and 9-6). The distribution of any accompanying sensory deficit similarly reflects the location of the underlying lesion (see Chapter 10, Sensory Disorders).
Table 9-5. Innervation of Selected Muscles of the Upper Limbs.
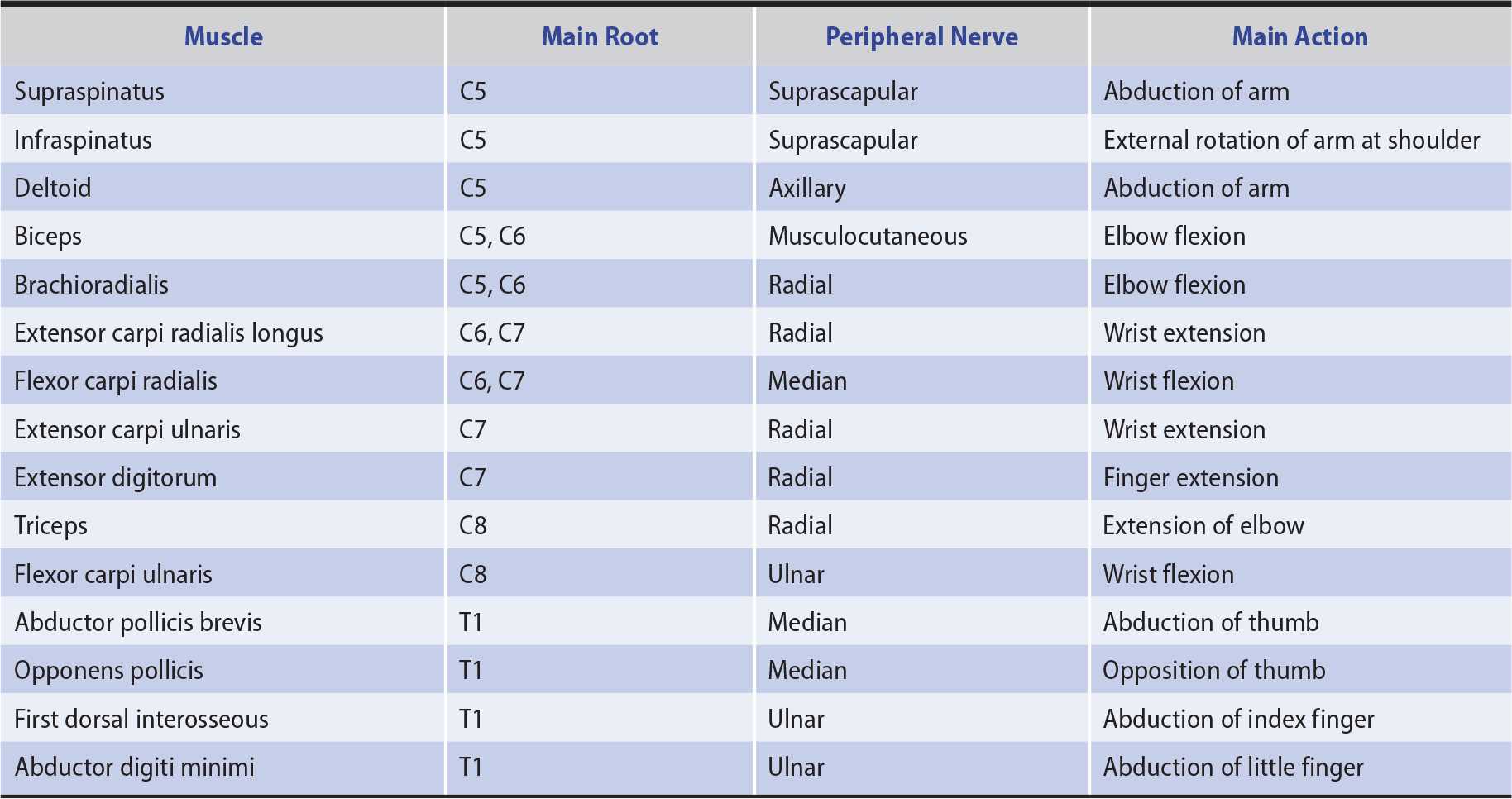
Table 9-6. Innervation of Selected Muscles of the Lower Limbs.
Weakness due to lesions of nerve roots may be difficult to distinguish from weakness caused by spinal cord lesions involving the anterior horn cells. In the latter situation, however, there is more often a bilateral motor deficit at the level of the lesion, a corticospinal or sensory deficit below it, or a disturbance of bladder, bowel, or sexual function.
Disorders affecting the anterior horn cells of the spinal cord commonly cause an extensive lower motor neuron deficit without sensory changes, and this helps to distinguish them from disorders of motor nerves (motor neuropathy).
CEREBELLAR LESIONS
 Signs
Signs
1. Hypotonia.
2. Depressed or pendular tendon reflexes.
3. Ataxia—This is a complex movement disorder caused, at least in part, by impaired coordination. It occurs in the limbs on the same side as a lesion affecting the cerebellar hemisphere. With midline lesions, incoordination may not be evident in the limbs, but there is marked truncal ataxia on walking. The term dysmetria is used when movements are not adjusted accurately for range, so that, for example, a moving finger overshoots a target at which it is aimed. Dysdiadochokinesia denotes rapid alternating movements that are clumsy and irregular in terms of rhythm and amplitude. Asynergia or dyssynergia denotes the breakdown of complex actions into the individual movements composing them; when asked to touch the tip of the nose with a finger, for example, the patient may first flex the elbow and then bring the hand up to the nose instead of combining the maneuvers into one action. Intention tremor occurs during activity and is often most marked as the target is neared. The rebound phenomenon is the overshooting of the limb when resistance to a movement or posture is suddenly withdrawn.
4. Gait disorder—The gait becomes unsteady in patients with disturbances of either the cerebellar hemispheres or midline structures.
5. Imbalance of station.
6. Disturbances of eye movement—Jerk nystagmus, which is commonly seen in patients with a unilateral lesion of the cerebellar hemisphere, is slowest and of greatest amplitude when the eyes are turned to the side of the lesion. Nystagmus is not present in patients with lesions of the anterior cerebellar vermis.
7. Dysarthria—Speech becomes dysarthric and takes on an irregular and explosive quality in patients with lesions involving the cerebellar hemispheres. Speech is usually unremarkable when only the midline structures are involved.
 Localization of the Underlying Lesion
Localization of the Underlying Lesion
The relationship of symptoms and signs to lesions of different parts of the cerebellum is considered in Chapter 8, Disorders of Equilibrium.
NEUROMUSCULAR TRANSMISSION DISORDERS
 Signs
Signs
1. Normal or reduced muscle tone.
2. Normal or depressed tendon and superficial reflexes.
3. No sensory changes.
4. Weakness, often patchy in distribution, not conforming to the distribution of any single anatomic structure; frequently involves the cranial muscles and may fluctuate in severity over short periods, particularly in relation to activity.
 Localization of the Underlying Lesion
Localization of the Underlying Lesion
Pathologic involvement of either the presynaptic (eg, botulism) or postsynaptic (eg, myasthenia gravis) portion of the neuromuscular junction may impair neuromuscular transmission. Disorders of neuromuscular transmission are discussed later.
MYOPATHIC DISORDERS
 Signs
Signs
1. Weakness, usually most marked proximally rather than distally.
2. No muscle wasting or depression of tendon reflexes, at least until an advanced stage of the disorder.
3. Normal abdominal and plantar reflexes.
4. No sensory loss or sphincter disturbances.
 Localization of the Underlying Lesion
Localization of the Underlying Lesion
It It is important to determine whether the weakness is congenital or acquired, whether there is a family history of a similar disorder, and whether there is any clinical evidence that a systemic disease may be responsible. The distribution of affected muscles is often especially important in distinguishing the various hereditary myopathies (see Myopathic Disorders, later).
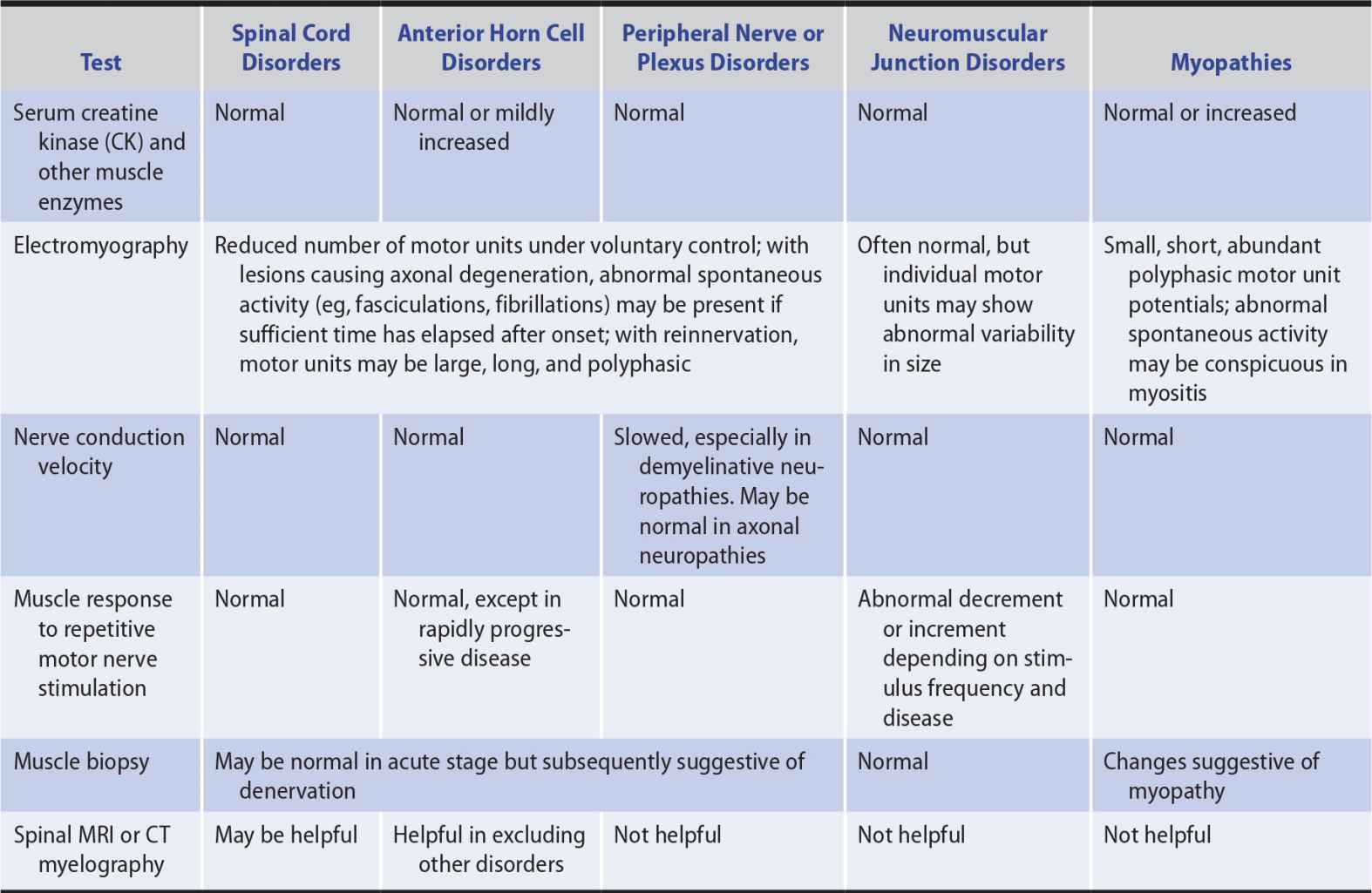
INVESTIGATIVE STUDIES
Investigative studies of patients with weakness from focal cerebral deficits are considered in Chapter 2, Investigative Studies. The investigations discussed here may be helpful in evaluating patients with weakness from other causes (Table 9-7).
Table 9-7. Investigation of Patients With Weakness of Noncerebral Origin.
IMAGING
 Plain X-Rays of the Spine
Plain X-Rays of the Spine
Congenital abnormalities and degenerative, inflammatory, neoplastic, or traumatic changes may be revealed by plain x-rays of the spine, but magnetic resonance imaging (MRI) or computed tomography (CT) scan is preferred in patients with suspected cord or root lesions because it provides more detail and visualization of soft tissue structures.
 CT Scan or MRI
CT Scan or MRI
CT scan of the spine, especially after instilling water-soluble contrast material into the subarachnoid space (CT myelogram), may reveal disease involving the spinal cord or nerve roots, but MRI is better in this regard (see Chapter 2, Investigative Studies).
ELECTRODIAGNOSTIC STUDIES
The function of the normal motor unit, which consists of a lower motor neuron and all of the muscle fibers it innervates, may be disturbed at any of several sites in patients with weakness. A lesion may, for example, affect the anterior horn cell or its axon, interfere with neuromuscular transmission, or involve the muscle fibers directly so that they cannot respond normally to neural activation. In each circumstance, characteristic changes in the electrical activity can be recorded from affected muscle by a needle electrode inserted into it and connected to an oscilloscope (electromyography, or EMG). Depending on the site of pathology, nerve conduction studies or the muscle responses to repetitive nerve stimulation may also be abnormal (see Table 9-7 and Chapter 2, Investigative Studies, for further details).
SERUM ENZYMES
Damage to muscle fibers may lead to the release of certain enzymes (creatine kinase [CK], aldolase, lactic acid dehydrogenase [LDH], and alanine and aspartate transaminases [ALT and AST]) that can then be detected in increased amounts in serum. Serum CK shows the greatest increase and is the most useful for following the course of muscle disease. It is also present in high concentrations in the heart and brain, however, and damage to these structures can lead to increased serum CK levels. Fractionation of serum CK into isoenzyme forms is useful for determining the tissue of origin. In patients with weakness, elevated serum CK levels are generally indicative of a primary myopathy, especially one that is evolving rapidly. A moderately elevated serum CK may also occur in motor neuron disease, however, and more marked elevations can follow trauma, surgery, intramuscular injections, EMG, or vigorous activity.
MUSCLE BIOPSY
Histopathologic examination of a specimen of weak muscle can help to determine whether the underlying weakness is neurogenic or myopathic. With neurogenic disorders, atrophied fibers occur in groups, with adjacent groups of larger, uninvolved fibers. In myopathies, atrophy occurs in a random pattern; nuclei of muscle cells may be centrally situated, in contrast to their normal peripheral location; and fibrosis or fatty infiltration may be seen. In addition, the pathologic examination may permit recognition of certain inflammatory muscle diseases (eg, polymyositis) for which specific treatment is available and of various congenital or mitochondrial myopathies.
SPINAL CORD DISORDERS (MYELOPATHY)
Spinal cord disorders can lead to motor, sensory, or sphincter disturbances, or to some combination of these deficits. Depending on whether it is unilateral or bilateral, a lesion above C5 may cause an ipsilateral hemiparesis or quadriparesis. With lesions lower in the cervical spinal cord, involvement of the upper limbs is partial, and a lesion below T1 affects only the lower limbs on one or both sides. Disturbances of sensation are considered in detail in Chapter 10, Sensory Disorders, but unilateral involvement of the posterior columns of the cord leads to ipsilateral loss of position and vibration sense. Involvement of the spinothalamic tracts in the anterolateral columns impairs contralateral pain and temperature appreciation below the level of the lesion.
Spasticity is a common accompaniment of upper motor neuron lesions and may be especially troublesome below the level of a myelopathy. When the legs are weak, the increased tone of spasticity may help to support the patient in the upright position. Marked spasticity, however, may lead to deformity, interfere with toilet functions, and cause painful flexor or extensor spasms. Pharmacologic management includes treatment with diazepam, baclofen, dantrolene, or tizanidine, as discussed later under Traumatic Myelopathy, but reduction in tone may increase disability from underlying leg weakness.
TRAUMATIC MYELOPATHY
Although spinal cord damage may result from whiplash (recoil) injury, severe injury to the cord usually relates to fracture-dislocation in the cervical, lower thoracic, or upper lumbar region, which is commonly associated with local pain. Concomitant cerebral and systemic injuries may complicate evaluation. The most common site for traumatic spinal cord injury is in the cervical region.
CLINICAL FINDINGS
 Total Cord Transection
Total Cord Transection
Total transection results in immediate permanent paralysis and loss of sensation below the level of the lesion. Reflex activity is lost for a variable period, but then increases.
1. In the acute stage (“spinal shock”), there is flaccid paralysis with loss of tendon and other reflexes, accompanied by sensory loss and by urinary and fecal retention.
2. Over the following weeks, as reflex function returns, a spastic paraplegia or quadriplegia emerges, with brisk tendon reflexes and extensor plantar responses; however, a flaccid, atrophic (lower motor neuron) paralysis may affect muscles innervated by spinal cord segments at the level of the lesion, where anterior horn cells are damaged. Sensation is reduced at that level and lost below it. The bladder and bowel regain some reflex function, so that urine and feces are expelled at intervals.
3. Flexor or extensor spasms of the legs may become increasingly troublesome and are ultimately elicited by even the slightest cutaneous stimulus, especially in the presence of bedsores or a urinary tract infection. Eventually, the patient assumes a posture with the legs in flexion or extension, the former being especially likely with cervical or complete cord lesions.
 Less Severe Injury
Less Severe Injury
With lesser injury, the neurologic deficit is less severe, but patients may be left with a mild paraparesis or quadriparesis and/or a distal sensory disturbance. Sphincter function may also be impaired—urinary urgency and urgency incontinence are especially common. Hyperextension injuries of the neck can lead to focal cord ischemia that causes bibrachial paresis (weakness of both arms) with sparing of the legs and variable sensory signs.
IMAGING
Plain radiographs will reveal misalignments, fractures, and soft tissue swelling, but CT scanning is more sensitive for detecting spinal fractures, especially in the cervical region, and also allows evaluation of the spinal cord. It is therefore preferred in the acute setting. Spinal MRI provides complementary information about the extent and nature of any spinal cord injury and the presence of an epidural hematoma, which is important for treatment and prognosis.
TREATMENT
 Immobilization, Decompression, & Stabilization
Immobilization, Decompression, & Stabilization
Initial treatment consists of immobilization until the nature and extent of the injury are determined. Injury of the spinal cord should be assumed in patients with head injuries until excluded by imaging studies. If there is cord compression, urgent decompressive surgery will be necessary. An unstable spine may require surgical fixation, and vertebral dislocation may necessitate spinal traction.
 General Measures
General Measures
A clear airway must be ensured and the circulation, blood pressure, and ventilation maintained. Tracheostomy may be required. Respiratory complications such as pneumonia, atelectasis, and pulmonary embolism must be treated vigorously. Prophylaxis for deep venous thrombosis with low-molecular-weight heparin is important.
 Corticosteroids
Corticosteroids
It is doubtful whether corticosteroids (eg, methylprednisolone 30 mg/kg by intravenous bolus followed by intravenous infusion at 5.4 mg/kg/h for 24 hours), once thought to improve function at 6 months when begun within 8 hours of traumatic spinal cord injury, have any significant beneficial effects.
 Treatment of Painful Spasms
Treatment of Painful Spasms
Painful flexor or extensor spasms can be treated with drugs that enhance spinal inhibitory mechanisms (baclofen, diazepam) or uncouple muscle excitation from contraction (dantrolene). Baclofen is started with 5 mg orally twice daily, increasing up to 30 mg four times daily; diazepam, 2 mg orally twice daily up to as high as 20 mg three times daily; and dantrolene, 25 mg/d orally to 100 mg four times daily. Tizanidine, a central α2-adrenergic receptor agonist, may also be helpful by increasing presynaptic inhibition and reducing alpha motoneuron excitability. The daily dose is built up gradually, usually to 8 mg three times daily. Side effects include dryness of the mouth, somnolence, and hypotension, but the drug is usually well tolerated. Patients who fail to benefit from or who cannot tolerate sufficient doses of oral medications may respond to intrathecal infusion of baclofen.
All these drugs may increase functional disability by reducing tone. Dantrolene may also increase weakness and should be avoided in patients with severely compromised respiratory function.
 Skin Care
Skin Care
Skin care is important; continued pressure on any single area must be avoided.
 Bladder & Bowel Disorders
Bladder & Bowel Disorders
Depending on the severity of injury, catheterization may be necessary initially. Subsequently, the urgency and frequency of the spastic bladder may respond to a parasympatholytic drug such as oxybutynin, 5 mg three times daily. Suppositories and enemas will help maintain regular bowel movements and may prevent or control fecal incontinence.
 Experimental Therapeutics
Experimental Therapeutics
Experimental work has focused recently on enhancing axonal regeneration in the damaged section of the spinal cord through such approaches as neutralization of neurite regrowth inhibitors, use of neurotrophic or growth factors, implantation of synthetic axonal guidance channels, and cellular therapies. Translational application to patients with spinal cord injuries is possible in the near future.
PROGNOSIS
There is a significant mortality after spinal cord injury, and this is highest in those with cervical injuries, associated head injury, cardiovascular or respiratory inadequacy, and coexisting disorders. The greatest improvement is seen in those with incomplete injury, with most recovery occurring in the first few months.
DEMYELINATING MYELOPATHIES
MULTIPLE SCLEROSIS
 Epidemiology
Epidemiology
Multiple sclerosis is one of the most common neurologic disorders, affecting approximately 300,000 patients in the United States, and its highest incidence is in young adults. It is defined clinically by the involvement of different parts of the central nervous system at different times—provided that other disorders causing multifocal central dysfunction have been excluded. Initial symptoms generally commence before the age of 55 years, with a peak incidence between ages 20 and 40 years; women are affected nearly twice as often as men.
Epidemiologic studies show that the prevalence of the disease rises with increasing distance from the equator, and no population with a high risk for the disease exists between latitudes 40°N and 40°S. Vitamin D level may also play a role as may exposure to Epstein-Barr virus. A genetic predisposition is suggested by twin studies, the occasional familial occurrence, and the strong association between the disease and specific HLA alleles. Alleles of IL2RA (interleukin-2 receptor α gene) and IL7RA (interleukin-7 receptor α gene) have also been proposed as heritable risk factors. Present evidence suggests that the disease has an autoimmune basis.
 Pathology
Pathology
The disorder is characterized pathologically by the development of focal—often perivenular—scattered areas of demyelination, together with reactive gliosis, axonal damage, and neuronal degeneration. These lesions occur in both white and gray matter of the brain and spinal cord and in the optic (II) nerve.
 Pathophysiology
Pathophysiology
The cause of multiple sclerosis is unknown, but tissue damage and neurologic symptoms are thought to be triggered by an immune mechanism directed against myelin antigens. Viral infection or other inciting factors may promote the entry of T cells and antibodies into the central nervous system by disrupting the blood–brain barrier. This leads to increased expression of cell-adhesion molecules, matrix metalloproteinases, and proinflammatory cytokines. These molecules work in concert to attract additional immune cells, break down the extracellular matrix to aid their migration, and activate autoimmune responses against several antigens (eg, myelin basic protein, myelin-associated glycoprotein, myelin oligodendrocyte glycoprotein, proteolipid protein, α B-crystallin, phosphodiesterases, and S-100). Binding of these target antigens by antigen-presenting cells triggers an autoimmune response that may involve cytokines, macrophages, and complement. Immune attack on myelin denudes axons, which slows nerve conduction. Together with loss of axons and nerve cell bodies, this leads to progressive neurologic symptoms.
 Clinical Findings
Clinical Findings
1. Initial or presenting symptoms—Patients can present with any of a variety of symptoms (Table 9-8). Common initial complaints are focal weakness, numbness, tingling, or unsteadiness in a limb; sudden loss or blurring of vision in one eye (optic neuritis); diplopia; disequilibrium; or a bladder-function disturbance (urinary urgency or hesitancy). Symptoms are often transient, disappearing after a few days or weeks, even though some residual deficit may be found on neurologic examination. Other patients present with an acute or gradually progressive spastic paraparesis and sensory deficit; this should raise concern about the possibility of an underlying structural lesion unless there is evidence on clinical examination of more widespread disease.
2. Subsequent course—Months or years may elapse after the initial episode before further symptoms appear. Then, either new symptoms develop or the original ones recur and progress. Relapses may be triggered by infection and, in women, are more likely in the 3 months or so after childbirth (but are reduced during the pregnancy itself). A rise in body temperature can cause transient deterioration in patients with a fixed and stable deficit (Uhthoff phenomenon). With time—and after a number of relapses and usually incomplete remissions—the patient may become increasingly disabled by weakness, stiffness, sensory disturbances, unsteadiness of the limbs, impaired vision, and urinary incontinence.
Table 9-8. Symptoms and Signs of Multiple Sclerosis.
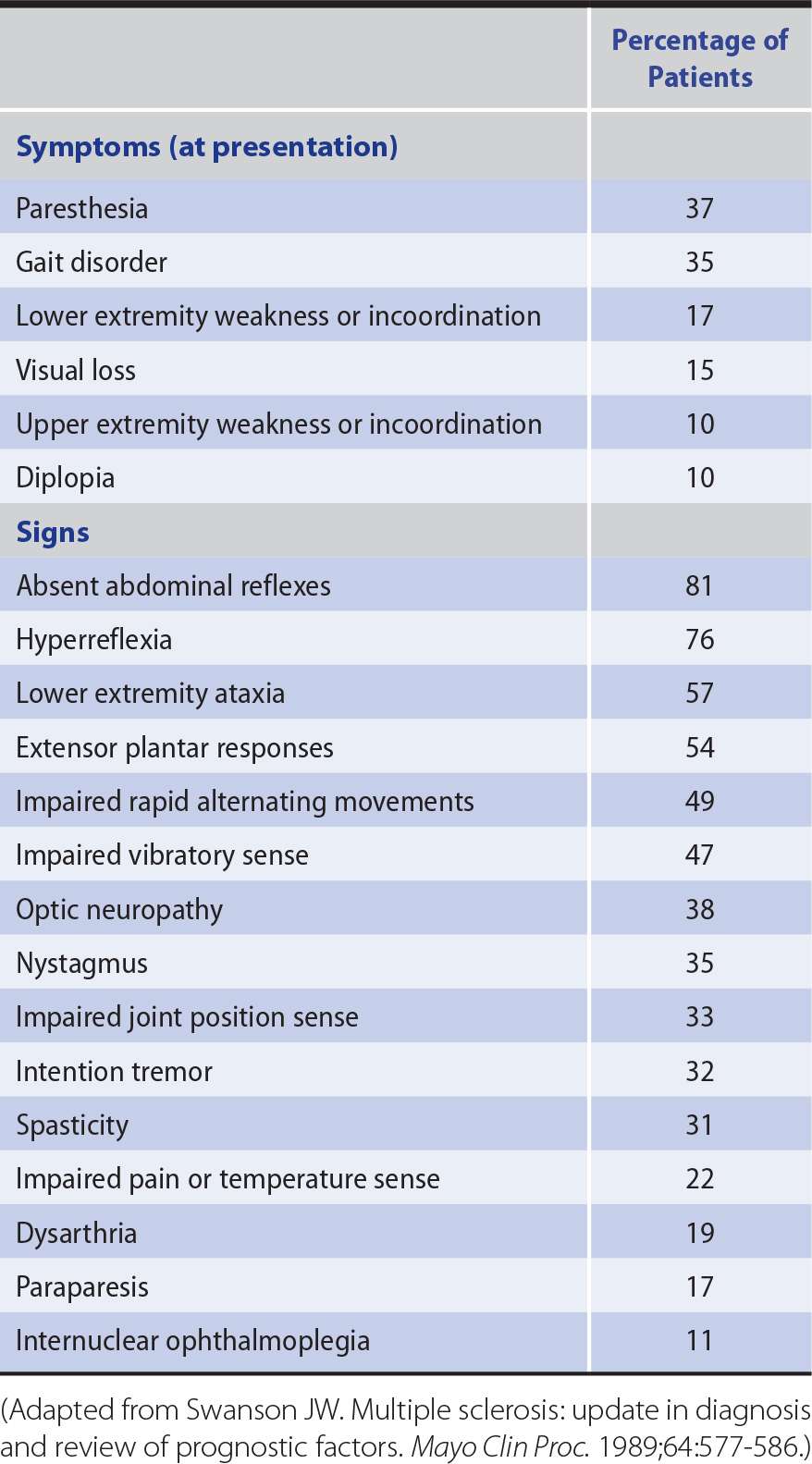
Based on its course, the disease is divided into a relapsing-remitting form (85% of cases), in which progression does not occur between attacks; a secondary progressive form (80% of cases after 25 years) characterized by a gradually progressive course after an initial relapsing-remitting pattern; and a primary progressive form (10% of cases) with gradual progression of disability from clinical onset. A progressive-relapsing form occurs rarely, with acute relapses being superimposed on a primary progressive course.
Examination in advanced cases commonly reveals optic atrophy, nystagmus, dysarthria, and upper motor neuron, sensory, or cerebellar deficits in some or all of the limbs (see Table 9-8). The diagnosis cannot be based on any single symptom or sign but only on a total clinical picture that indicates involvement of different parts of the central nervous system at different times.
 Investigative Studies
Investigative Studies
These may support the clinical diagnosis and exclude other disorders but do not themselves justify a definitive diagnosis of multiple sclerosis.
The cerebrospinal fluid (CSF) is commonly abnormal, with mild lymphocytosis or a slightly increased protein concentration, especially if examined soon after an acute relapse. CSF protein electrophoresis shows the presence of discrete bands in the immunoglobulin G (IgG) region (oligoclonal bands) in 90% of patients. The antigens responsible for these antibodies are not known.
If clinical evidence of a lesion exists at only one site in the central nervous system, a diagnosis of multiple sclerosis cannot properly be made unless other regions are affected subclinically. Such subclinical involvement may be detected by the electrocerebral responses evoked by monocular visual stimulation with a checkerboard pattern (visual evoked potentials), monaural stimulation with repetitive clicks (brainstem auditory evoked potentials), or electrical stimulation of a peripheral nerve (somatosensory evoked potentials).
MRI may also detect subclinical lesions and has become nearly indispensable in confirming the diagnosis (Figure 9-4). T1-weighted images may reveal hypointense “black holes” that probably represent areas of permanent axonal damage; hyperintense lesions are also found. Gadolinium-enhanced T1-weighted images may highlight areas of inflammation with breakdown of the blood–brain barrier. T2-weighed images provide information about disease burden or lesion load (ie, total number of lesions), which typically appear as areas of high signal intensity. Other MRI techniques, including measures of cerebral atrophy, magnetization transfer imaging, magnetic resonance spectroscopy, and diffusion tensor imaging, will likely provide yet more relevant information. The MRI of healthy subjects sometimes shows “unidentified bright objects” that resemble the lesions of multiple sclerosis but are without clinical correlates or significance; the imaging findings must therefore be interpreted in the clinical context in which they were obtained.
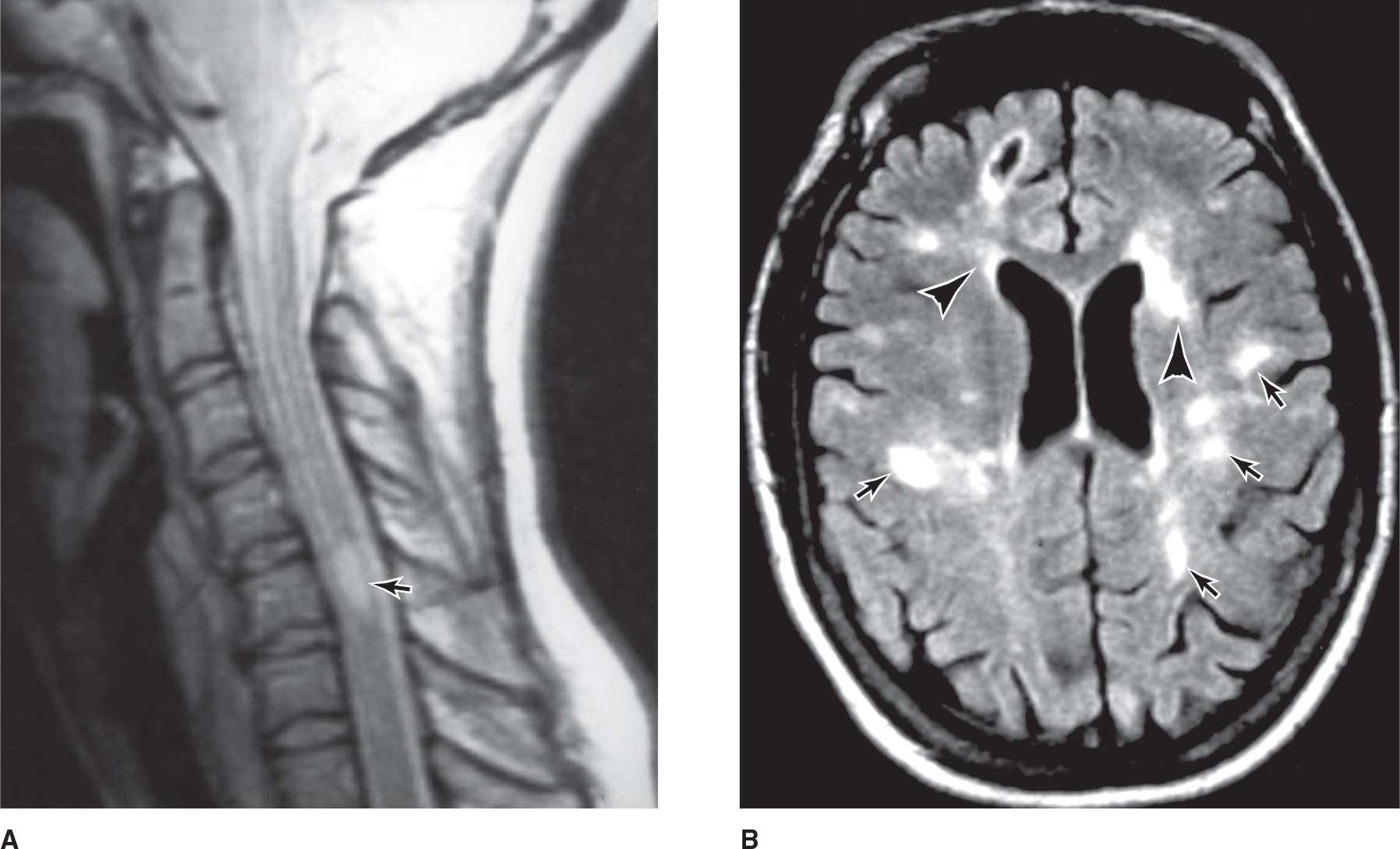
![]() Figure 9-4. (A) A mid-sagittal T2-weighted MRI of the cervical spinal cord in a young woman with multiple sclerosis. An abnormal region of high signal intensity (arrow) is seen. (Used with permission from RA Heyman.) (B) Axial T2-weighted MR brain image of a patient with multiple sclerosis showing multiple, primarily punctate, white matter plaques (arrows); note the typical location in the periventricular region (arrowheads). (Used with permission from RA. Heyman.)
Figure 9-4. (A) A mid-sagittal T2-weighted MRI of the cervical spinal cord in a young woman with multiple sclerosis. An abnormal region of high signal intensity (arrow) is seen. (Used with permission from RA Heyman.) (B) Axial T2-weighted MR brain image of a patient with multiple sclerosis showing multiple, primarily punctate, white matter plaques (arrows); note the typical location in the periventricular region (arrowheads). (Used with permission from RA. Heyman.)
Spinal MRI or CT myelography may be necessary to exclude a single congenital or acquired surgically treatable lesion in patients with spinal involvement and no evidence of disseminated disease. The region of the foramen magnum must be visualized to exclude the possibility of a lesion such as Arnold-Chiari malformation, in which part of the cerebellum and the lower brainstem are displaced into the cervical canal, producing mixed pyramidal and cerebellar deficits in the limbs.
 Diagnosis
Diagnosis
The diagnosis of multiple sclerosis requires evidence that at least two different regions of the central white matter have been affected at different times. Multiple sclerosis can be diagnosed straightaway in patients with at least two typical attacks and two MRI lesions. In a patient with only one lesion, a further attack involving a different site suffices to show dissemination in space; otherwise, repeat imaging in a few months should demonstrate at least one T2 lesion in at least two of four typical sites (periventricular, juxtacortical, infratentorial, or spinal). To show dissemination in time in a patient with only one attack, the simultaneous presence on MRI of asymptomatic gadolinium-enhancing and non-enhancing lesions at any time is sufficient; alternatively, it is necessary to await development of a new T2 or gadolinium-enhancing lesion on follow-up MRI or a second clinical attack. Diagnosis of primary progressive disease requires at least one year of progressive disease plus two of the following: (1) at least one typical T2 brain lesion, (2) at least two spinal T2 lesions, and (3) positive CSF oligoclonal bands, increased IgG index, or both.
In patients with only a single clinical event and who do not satisfy criteria for multiple sclerosis, a clinically isolated syndrome (CIS) is diagnosed. These patients are at increased risk for developing multiple sclerosis and are sometimes offered treatment as if they had the disease in the hope of delaying progression to clinically definite disease. Follow-up MRI should be considered 6 to 12 months later to determine whether any new lesions have occurred.
 Treatment
Treatment
The treatment approach is summarized in Table 9-9.
1. Relapsing-remitting disease—Treatment on an indefinite basis with interferon β-1a (30 μg intramuscularly once weekly, or 44 μg subcutaneously three times per week) or interferon β-1b (0.25 mg subcutaneously every other day) reduces the relapse rate. Glatiramer acetate (a mixture of random polymers simulating the amino acid composition of myelin basic protein) given by subcutaneous injection (20 mg daily) appears to be equally effective. In addition to their effect on relapses, interferon β-1a and glatiramer acetate may also delay the onset of significant disability in patients with relapsing disease. Natalizumab, an α4 integrin antibody, reduces the relapse rate when given intravenously once each month, but rarely has been associated with progressive multifocal leukoencephalopathy. It is given without other immune-modulating therapies to patients with relapsing-remitting disease poorly responsive to other therapies or with an aggressive initial course. However, if JC virus antibody testing is negative, the risk is low. Alemtuzumab, a lymphocyte inhibitor that also increases the risk of opportunistic infection, is approved in Europe but not the United States; its role in treatment remains undefined. Plasmapheresis is sometimes helpful when patients have severe relapses that are unresponsive to corticosteroids.
Interferons may cause a flu-like syndrome and (in the case of interferon β-1b) injection site reactions. Glatiramer acetate is generally tolerated well but may produce erythema at injection sites, and approximately 15% of patients experience transient episodes of flushing, dyspnea, chest tightness, palpitations, and anxiety after injections. All three of these agents are approved for use in relapsing-remitting multiple sclerosis. They are expensive, but their cost must be balanced against the reduced need for medical care and reduced time lost from work that follows their use.
Oral therapies are now available and are preferred by some, although their long-term safety profile is less clear. There are no trials comparing the efficacy of these newer agents. Fingolimod (0.5 mg daily) reduces relapses and disease progression; it also reduces MRI lesion activity and loss of brain volume in relapsing-remitting disease. Its mechanism of action is unknown but probably involves prevention of lymphocytes from migrating into the central nervous system. It is generally safe and well tolerated, but is contraindicated after recent myocardial infarction or with certain other cardiac disturbances. Adverse effects include headache, fatigue, back pain, diarrhea, respiratory tract infections, elevation of liver enzymes, blood pressure effects, macular edema, and—on initiation of therapy—transient bradycardia and slowed atrioventricular conduction. Therefore, heart rate should be monitored for 6 hours after the first dose, or if fingolimod is restarted after its use was interrupted for 2 weeks or more. Skin and certain other cancers have also been reported. At least until further experience has accumulated, use of fingolimod is probably best restricted to patients with active relapsing-remitting disease who are intolerant of β interferons and glatiramer acetate; it is also prescribed for newly diagnosed patients with active relapsing disease who prefer treatment with oral rather than parenteral medications, provided they understand the associated risks. Another oral agent, dimethyl fumarate (120 mg twice daily for 1 week, then 240 mg twice daily), also reduces relapse rate. Side effects include flushing, gastrointestinal complaints (eg, diarrhea, nausea, abdominal pain), and a reduced peripheral lymphocyte count. Relapse rate and disease progression is reduced also by teriflunomide, an immunomodulatory drug (taken orally in a daily dose of 7 or 14 mg). It has risks of hepatotoxicity and teratogenicity; side effects include alopecia, nausea, diarrhea, paresthesias, flu-like symptoms, elevated serum transaminases, and peripheral neuropathy. Corticosteroids may hasten recovery from acute relapses, but the extent of the recovery itself is unchanged. Treatment is therefore generally reserved for attacks that lead to acute change in functional ability, such as by causing visual or gait dysfunction. Long-term corticosteroid administration does not prevent relapses and should not be used because of unacceptable side effects. There is no standard schedule of treatment with corticosteroids, but the regimen most commonly used is intravenous methylprednisolone (1 g daily) for 5 days, followed by an oral prednisone taper (1 mg/kg/d for 1 week, with rapid reduction over the ensuing 1-2 weeks).
2. Primary or secondary progressive multiple sclerosis—Optimal treatment in these forms of the disease is less clear. There is no established treatment for primary progressive multiple sclerosis. Interferon β-1b (and probably interferon β-1a) are effective in reducing the progression rate as determined clinically and by MRI in secondary progressive disease, but there is only limited experience with glatiramer acetate in this setting. Mitoxantrone probably reduces the clinical attack rate and may help to reduce disease progression in patients whose clinical condition is worsening. Treatment with cyclophosphamide, azathioprine, or methotrexate, may help to arrest the course of secondary progressive disease, but studies are inconclusive. Pulse therapy with high-dose intravenous methylprednisolone (1 g/d once a month) is also sometimes effective and may carry a lower risk of long-term complications than the cytotoxic drugs. A recent phase 2 study suggests that simvastatin may have a role in treating secondary progressive disease.
3. General health and symptomatic treatment—Exercise and physical therapy are important, but excessive exertion must be avoided, particularly during periods of acute relapse. Fatigue is a serious problem for many patients and sometimes responds to amantadine or one of the selective serotonin reuptake inhibitor antidepressants. Treatment for spasticity (discussed earlier) is often needed, as is aggressive bladder and bowel management. Treatment for other aspects of advanced multiple sclerosis such as cognitive deficits, pain, tremor, and ataxia is generally less successful.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


