TRANSGENIC MOUSE MODELS OF AMYLOID-BETA AMYLOIDOSIS
Although human familial AD (FAD) comprises a small portion of AD cases, the identification of genetic mutations associated with FAD in genes coding for the amyloid precursor protein (APP) or presenilin 1 and 2 (PS1, PS2) led to development of the earliest tg mouse models. APP is cleaved by proteolytic enzymes, ß– and γ-secretases, to form amyloid-beta (Aß) 40 and 42, the main components of the amyloid plaques1. Mutations in the APP gene create excess Aß deposition by enhancing ß-secretase cleavage of APP or by altering the pattern of γ-secretase cleavage of APP. This results in a higher Aß42 to Aß40 ratio, which in turn enhances Aß fibrillogenesis.
One of the first strategies to create AD-like pathology in mice was to develop tg mouse models that over-express human APP with FAD mutations. While over-expression of APP is not a general feature of AD, this approach led to tg mice with significant Aß production and deposition over their lifetime. One such tg mouse model, known as PDAPP, uses a platelet-derived growth factor ß (PDGF ß) promoter to enhance in mice the expression of human APP with V717F mutation2. Another early tg mouse model, Tg2576, uses the hamster prion protein promoter (PrP ) to enhance the expression of human APP with a double Swedish mutation (K670N/M671L)3. Both models have been extensively studied as models of Aß amyloidosis (Table 44.1).
Another approach to developing tg AD mouse models involved the insertion of human presenilin genes with mutations known to cause AD into the mouse genome. Presenilins are part of the γ-secretase complex, which is involved in the cleavage of APP along with ß-secretase. However, mouse models with PS1 mutations alone do not develop amyloid pathology4,5. When both APP and PS1 mutations are expressed together in tg mice, such as in APPswe/PS1dE9 (Table 44.1)6, the PS1 mutation increases the ratio of Aß42 to Aß40, resulting in more Aß deposition due to the higher fibrillogenic nature of Aß427.
Many of the tg mouse models of amyloidosis also develop other pathologies seen in the human disease such as neuritic plaques, glio- sis or synaptic loss, allowing for observation of the effect of amyloid on neuronal pathologies. Significantly, monoaminergic neurodegeneration, a feature of the human disease, occurs in at least one mouse model8. These models have been very useful to the identification of mechanism-based therapeutics, specifically, anti-amyloid therapies. However, a major limitation is that the amyloidosis models do not develop NFTs, one of the hallmark neuropathological changes seen in AD.
TRANSGENIC MOUSE MODELS OF TAUOPATHY
The amyloid cascade hypothesis1 proposes that Aß is an early upstream agent that is followed by excess phosphorylation of tau, an intracellular, microtubule-associated protein. Hyperphosphorylation of tau is thought to lead to the formation of paired helical filaments (PHF), which progress to become NFTs in the neuronal cell body. However, as mentioned above, tg mouse models of amyloidosis do not form NFTs. One reason may be differences in tau isoforms across humans and mice. Six human isoforms of tau exist, each derived from alternative splicing in the brain of the same gene. There are only three isoforms in mice, which may result in a lower propensity for hyperphosphorylation9.
There is currently no known tau mutation in AD. However, independent of amyloid plaques, tau inclusions are found in other neurogenerative diseases such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease (PiD) and frontotemporal dementia (FTD). Therefore, tg mouse models of tauopathy have been created using human mutations in the tau
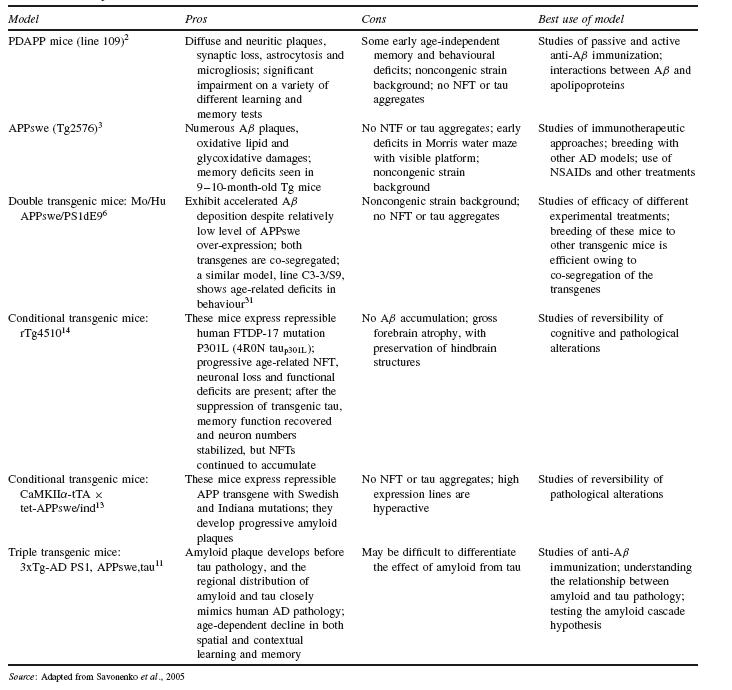
gene linked to familial frontotemporal dementia with Parkinsonism on chromosome 17 (FTDP-17). One such model, expressing the FTDP-17 mutation P301L, forms NFTs in diencephalon, brainstem, cerebellum and spinal cord. However, this model also has motor deficits due to spinal cord lesions such as fibrillary gliosis and axonal degeneration10.
More recently, models which reproduce both amyloid plaques and NFTs have been developed, the triple transgenic mice 3xTg-AD, which carry PS1 (M146V), APP (K670N/M671L) and tau (P301L) transgenes (Table 44.1)11. Interestingly, even though both APP and tau transgenes are expressed to similar levels in these mice, Aß deposits emerge prior to tau pathology, consistent with the amyloid cascade hypothesis. Immunotherapy experiments involving direct injection of anti-Aß antibodies into the hippocampus showed clearance of Aß deposits followed by clearance of earlier (non- hyperphosphorylated) forms of tau. Aß deposits re-emerged by 30 days post-injection, with tau pathology re-emerging at a later time point. Injection of anti-tau antibody had no observable effect on either Aß or tau deposits. The data from these experiments further support the view that Aß precedes tau pathology per the amyloid cascade hypothesis12. These triple transgenic models are therefore quite useful for studying the role of tau protein in neurodegenerative diseases, and especially its interaction with amyloid pathology.
CONDITIONAL TRANSGENIC MOUSE MODELS
Researchers have also developed variations of the above models in which the expression of APP, tau or both transgenes are temporally controlled. The approach used, commonly known as the ‘tet-off system,’ creates a tg model in which administration of a tetracycline analogue, such as doxycycline, turns off transcription of the transgene. Conditional tg models allow study of the effects of temporal variation (early vs. late, short vs. long) in the expression of the transgene. Such systems also model pharmaceutical interventions in which the expression of a transgene is suppressed, allowing assessment of potential treatment effect13. They can also test the hypothesis of whether certain structural and biochemical changes are related to the expression of the transgene14.
A well-known conditional APP tg mouse model is CaMKIIα- tTA × tet-APPswe/ind (Table 44.1). This model expresses amyloid plaques starting eight weeks of age, and amyloid deposition is severe by six months of age. APP expression is reduced by greater than 95% two weeks after initiation of doxycycline treatment. When doxycycline was given to examine the effects of Aß suppression after the onset of deposition, further progression of Aß deposition was halted, but there was limited to no clearance of previously deposited amyloid plaques even after six months of treatment. These results suggested that inhibition of Aß production may halt further progression of the pathology, but may not clear the pre-existing amyloid plaques13.
A tg mouse model with conditional tau expression is rTg4510, in which mutant tau (P301L) expression is suppressed by administration of doxycycline (Table 44.1
Related posts:
 The Care Home Experience Alisoun Milne
The Care Home Experience Alisoun Milne
 The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
 The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Depression After Stroke Peter Knapp and Allan House
Sleep and Ageing: Disorders and Management Helen Chiu and Joshua Tsoh
The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Depression After Stroke Peter Knapp and Allan House
Sleep and Ageing: Disorders and Management Helen Chiu and Joshua Tsoh
 Training Requirements for Old Age Psychiatrists in the UK Susan Mary Benbow and Aparna Prasanna
Training Requirements for Old Age Psychiatrists in the UK Susan Mary Benbow and Aparna Prasanna
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


