Chapter 68 Movement Disorders
Introduction
Movement disorders are syndromes characterized by impaired voluntary movement, the presence of involuntary movements, or both. There may be impaired targeting and velocity of intended movements, abnormal involuntary movements, abnormal postures, or excessive normal-appearing movements at inappropriate or unintended times. Movement disorders in children include athetosis, chorea, dystonia, myoclonus, parkinsonism, stereotypies, tics, and tremor [Sanger, 2003a]. Movement disorders may be accompanied by weakness, spasticity, hypotonia, ataxia, apraxia [Koski et al., 2002], and other motor deficits, although many authors do not include these accompanying deficits.
Movement disorder terminology has been well defined for adults, but less so for children. Therefore, it is likely that movement disorders are under-reported in children, and that there is inconsistent terminology. Recently, there have been attempts to provide specific definitions of childhood motor disorders [Sanger et al., 2003, 2006, 2010]. According to these definitions, childhood disorders can be divided into three major categories: hypertonic disorders, hyperkinetic disorders, and negative signs. Hypertonic disorders include spasticity, dystonia, and rigidity. Hyperkinetic disorders include chorea, dystonia, athetosis, myoclonus, tremor, stereotypies, and tics. Negative signs include weakness, reduced selective motor control, ataxia, apraxia, and developmental dyspraxia, although we will not discuss these here. Consensus definitions for these terms have been established, although the list is not intended to be exhaustive and disorders of gait, balance, speech, and eye movement are not included. Several important terms, including hypotonia and bradykinesia, have not yet been defined for children. The prevalence in children of different types of disorders is not known, although there have been studies investigating symptoms in certain populations, including children with cerebral palsy.
Characteristic Features of Pediatric Movement Disorders
Movement disorders in children differ from those in adults in several aspects. Perhaps the most important is that movement disorders in childhood are primarily symptoms of other diseases, rather than diseases in themselves [Sanger, 2003a, b]. In adults, dystonia and parkinsonism are usually due to primary dystonia or idiopathic Parkinson’s disease, respectively. However, dystonia or parkinsonism in children are more likely to be features of an underlying static or progressive neurological disorder. Diagnosis in children is complicated by the fact that many symptoms have more than one cause, and any particular underlying pathophysiology may lead to a complex combination of symptoms. The diagnostic work-up in children is guided by symptoms, but the existence of a large class of diseases that can lead to the same set of symptoms often necessitates a broad etiologic work-up. There may be both specific etiologic treatments, as well as symptomatic treatments, both of which may be beneficial in an individual child. In particular, many of the causes of childhood movement disorders do not yet have any specific treatment, yet symptomatic treatment for the resulting movement disorder can be extremely helpful and lead to improvement in quality of life.
Diagnosis of Movement Disorders
Classification of a movement disorder based upon the spatial and temporal pattern is essential for diagnosis. It is also important to define the context in which the movements occur. While it is often helpful to list the characteristics of the movements (Table 68-1), the diagnosis relies on pattern recognition, and the clinician must see the movements. If the movements are not apparent during the neurological examination, repeating the examination at another time or obtaining video recordings of the movements is important to making an accurate diagnosis. The widespread availability of video cameras has substantially improved diagnosis of movement disorders.
Table 68-1 Phenomenological Classification of Movement Disorders
| Movement Disorder | Brief Description |
|---|---|
| Athetosis | Slow, continuous, writhing movements of distal body parts, especially the fingers and hands |
| Chorea/ballism | Chaotic, random, repetitive, brief, purposeless movements. Rapid, but not as rapid as myoclonus. When very large in amplitude and affecting proximal joints, choreic limb movements are often called ballism |
| Dystonia | Repetitive, sustained, abnormal postures and/or movements. Abnormal postures typically have a twisting quality |
| Myoclonus | Sudden, brief, shocklike movements that may be repetitive or rhythmic |
| Parkinsonism | Hypokinetic syndrome characterized by a combination of rest tremor, slow movement (bradykinesia), rigidity, and postural instability |
| Stereotypy | Patterned, episodic, repetitive, purposeless, rhythmic movements |
| Tics | Stereotyped intermittent, sudden, discrete, repetitive, nonrhythmic movements, most frequently involving head and upper body |
| Tremor | Rhythmic oscillation about a central point or position, involving one or more body parts |
When approaching a patient with a movement disorder, it is helpful to address some key questions:
The Role of the Basal Ganglia in Movement Disorders
The basal ganglia are subcortical structures comprising several interconnected nuclei in the forebrain, midbrain, and diencephalon (Figure 68-1). They include the striatum (caudate, putamen, nucleus accumbens), the subthalamic nucleus (STN), the globus pallidus (internal segment [GPi]); external segment [GPe]; and ventral pallidum), and the substantia nigra (pars compacta [SNpc] and pars reticulata [SNpr]). The striatum and STN are the primary input structures of the basal ganglia, receiving excitatory input from cerebral cortex. The GPi and SNpr are the primary output nuclei, sending inhibitory output to thalamus and brainstem targets. Acting through the thalamus, the basal ganglia output influences frontal lobe cortical neurons. By virtue of the inhibitory output from the basal ganglia, conditions associated with destruction of the output nuclei may be associated with unwanted and nonspecific overactivity of thalamocortical and brainstem targets.
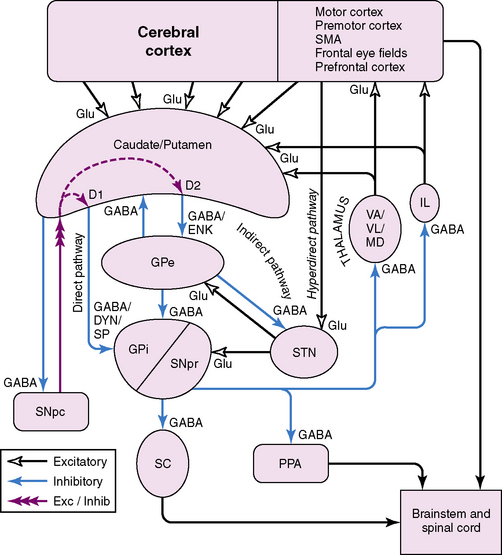
Medium spiny striatal neurons contain the inhibitory neurotransmitter GABA and are inhibitory to their targets. In addition, they have peptide neurotransmitters that are co-localized with GABA. Based on the type of neurotransmitters and the predominant type of dopamine receptor they contain, the medium spiny neurons can be divided into two populations. One population contains GABA, dynorphin, and substance P, and primarily expresses D1 dopamine receptors. These neurons project to the basal ganglia output nuclei, GPi, and SNpr, and form the “direct pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990]. The second population contains GABA and enkephalin, and primarily expresses D2 dopamine receptors. These neurons project to GPe and form the first limb of the “indirect pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990].
The STN receives an excitatory glutamatergic input from many areas of frontal lobes with particularly large inputs from motor areas of cortex. The STN also receives an inhibitory GABA input from GPe. The output from the STN is glutamatergic and excitatory to the basal ganglia output nuclei, GPi and SNpr. This projection forms the second limb of the “indirect pathway” [Alexander and Crutcher, 1990; Albin et al., 1989; DeLong, 1990].
According to the most common model of basal ganglia function, there are several distinct routes through which information flows from the cerebral cortex to the basal ganglia output nuclei. The two most direct are the disynaptic, inhibitory, “direct pathway” from cortex to striatum to GPi and SNpr, and the disynaptic, excitatory, “hyperdirect pathway” [Nambu et al., 2000] from cortex to STN to GPi and SNpr. The “direct pathway” is inhibitory to GPi and SNpr; the “hyperdirect pathway” is excitatory to GPi and SNpr. The “hyperdirect” pathway is the fastest route through the basal ganglia. There are several indirect routes, but the most important is the “indirect pathway” from cortex to striatum to GPe to STN to GPi and SNpr. Organization of these pathways confers a pattern of fast, powerful, and relatively broad excitation via the hyperdirect pathway, and more focused inhibition via the direct pathway (Figure 68-2) [Nambu et al., 2000; Parent and Hazrati, 1993; Mink, 1996]. The indirect pathway confers additional focus.
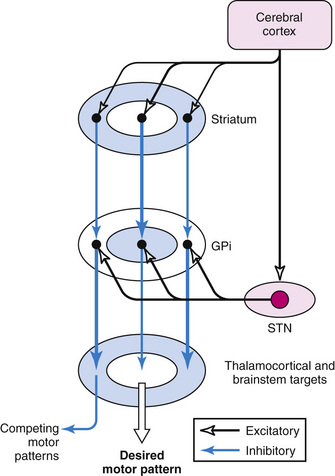
Output from basal ganglia to thalamocortical circuits appears to be segregated anatomically and possibly functionally. Thalamic targets of GPi and SNpr project, in turn, to frontal lobe, with the strongest output going to motor areas. The basal ganglia motor circuit has a somatotopic organization, with separate representation of different body parts maintained throughout the basal ganglia. There also appears to be relative segregation of motor from nonmotor basal ganglia circuits [Hoover and Strick, 1993].
A popular model of basal ganglia dysfunction in movement disorders was developed in the late 1980s (Figure 68-3) [Albin et al., 1989, 1995; DeLong, 1990]. In simple terms, the model proposes that hypokinetic disorders (e.g., parkinsonism) can be distinguished from hyperkinetic movement disorders (e.g., chorea, dystonia, tics), based on the magnitude and pattern of the basal ganglia output neurons in GPi and SNpr [Wichmann and DeLong, 1996]. Because basal ganglia output neurons are inhibitory to thalamus and the PPA, their function is analogous to a braking mechanism, such that increased activity inhibits and decreased activity facilitates motor pattern generators in cerebral cortex and brainstem [Mink, 1996]. As described above, the anatomic organization of the basal ganglia confers a pattern of focused facilitation and surround inhibition of motor mechanisms in thalamocortical and brainstem circuits (see Figure 68-2). The normal function of this organization is selectively to facilitate desired movements and to inhibit potentially competing movements [Mink, 1996, 2003].

Basal Ganglia Pathophysiology in Movement Disorders
Lesions in the striatum produce variable results that depend in part on the location of the lesion, in part on the lesion mechanism, and in part on what is measured. Lesions in the caudate nucleus most commonly cause behavioral disturbance, such as abulia, but may also cause chorea or dystonia. Lesions in the putamen are likely to produce movement deficits and typically cause dystonia (most common) or parkinsonism [Bhatia and Marsden, 1994].
A unilateral STN lesion is the classic cause of hemiballism, but smaller-amplitude chorea can also result from STN lesions [Carpenter and Carpenter, 1951]. When bilateral, STN lesions cause bilateral chorea or ballism. Most commonly, the chorea or hemiballism resulting from STN lesions involves the lower extremities more than the upper extremities, and can persist for days to months. The intensity of hemiballism usually diminishes over time. Globus pallidus lesions usually involve GPe and GPi, GPe and putamen, or GPi and SNpr. Such lesions can cause dystonia, parkinsonism, or both [Mink, 1996; Bhatia and Marsden, 1994; Heidenreich et al., 1988]. Chorea rarely accompanies globus pallidus lesions. Focal lesions of SNpr can cause involuntary eye movements [Hikosaka and Wurtz, 1985]. Lesions of SNpc dopamine neurons cause depletion of dopamine in the striatum, resulting in parkinsonism, dystonia, or both [Calne et al., 1997].
Focal basal ganglia lesions are uncommon in children. Movement disorders in children are more likely to result from global dysfunction of basal ganglia circuits. The model shown in Figure 68-3 has been used to explain the basis for hypokinetic and hyperkinetic movement disorders, but does not distinguish among the different hyperkinetic movement disorders. However, based on what is known about these disorders and the underlying neural circuitry, specific models have been proposed [Mink, 2003]. Diseases affecting the basal ganglia cause movement disorders that can be understood as failure to facilitate desired movements (e.g., Parkinson’s disease), failure to inhibit unwanted movements (e.g., chorea, dystonia, tics), or both [Albin et al., 1989; DeLong, 1990; Mink, 1996, 2003]. The involuntary movements of chorea, dystonia, and tics differ in important spatial and temporal characteristics that reflect important pathophysiologic differences. More extensive discussion of the neural basis of these disorder is contained in Mink [2003].
Etiology of Movement Disorders in Children
The causes of pediatric movement disorders are extensive. The most common cause of secondary disorders is likely to be cerebral palsy, with a prevalence of 2 per 1000. However, cerebral palsy itself represents a constellation of injuries and symptoms, and there is a wide range of types of injury, localization, and combinations of symptoms [SCPE, 2000]. Cerebral palsy can be associated with almost all forms of childhood movement disorders, and despite the lack of an on-going destructive process, the clinical picture may change during development. The diagnosis and management of cerebral palsy is complex and is discussed in Chapter 69.
Specific types of movement disorder may represent dysfunction of particular localized regions of the central nervous system [Sanger, 2003b]. Ataxia most likely occurs due to injury to the cerebellum or its afferent and efferent pathways [Taroni and DiDonato, 2004]. Bradykinesia most likely occurs with injury to the substantia nigra or striatum, leading to presynaptic or postsynaptic failure of dopaminergic transmission [Albin et al., 1989]. Chorea typically occurs with injury to the subthalamic nucleus, but it also can occur with widespread cerebral injury (e.g., following encephalitis). Dystonia most likely involves injury to the basal ganglia, but cortical or cerebellar abnormalities cannot be excluded as contributors [Hallett, 1998; Sanger, 2003b]. Myoclonus most likely involves cortical, brainstem, or spinal injury to gray matter [Caviness and Brown, 2004]. Localization of tremor depends upon the type, with some forms involving cerebellar or brainstem circuits [Uddin and Rodnitzky, 2003; Jankovic et al., 2004]. Tic disorders probably involve an abnormality of the basal ganglia, but cortical mechanisms may also contribute [Mink, 2001a].
Classification of Childhood Movement Disorders
The first step toward diagnosing and treating a movement disorder is to define the disorder (see Table 68-1). Individual names of movement disorders can refer to neurological signs or to neurological syndromes or diseases, which can cause some confusion. In this section, we will limit the definitions to the neurological signs to which they refer. In subsequent sections, we will present a more complete discussion of the syndromes and diseases in which the different signs are seen, either alone or in combination with other signs.
The most common movement disorders in the pediatric population are tic disorders. The prevalence of tic disorders in schoolchildren may be as high as 25 percent [Snider et al., 2002]. Most tic disorders are not severe, and the level of morbidity is low. Nevertheless, because of the lack of association with other neurological symptoms, it is not unreasonable to consider tic disorders as a primary disorder in idiopathic cases. There are rare exceptions in which tics are due to secondary disorders, such as brain injury or neuroacanthocytosis.
Dystonia is a relatively prevalent disorder in children. This most likely is due to its occurrence in cerebral palsy. Dystonia as the primary symptom in cerebral palsy is less common, but dystonia as a complicating factor, particularly in the upper extremities of children with cerebral palsy otherwise characterized by weakness or spasticity, is probably more common. The incidence and classification of cerebral palsy are discussed in Chapter 69.
Choreoathetosis is a term that has been applied to a specific subset of children with a dyskinetic form of cerebral palsy. This term is not generally used in adults. It is not known whether or not choreoathetosis represents a form of dystonia, chorea, or an entirely different disorder [Turny et al., 2004], but a recent consensus definition [Sanger et al., 2010] suggests that it is most appropriately considered to be a combination of chorea (random-appearing, more rapid movements) and athetosis (slower, flowing movements without intervening periods of rest). Certainly, choreoathetotic cerebral palsy (also commonly termed dyskinetic cerebral palsy) is an easily recognized syndrome, and since many of these children also possess some degree of dystonia as well as chorea, choreoathetosis may simply be the expression of a combination of other movement disorders [Morris et al., 2002a]. Spasticity and weakness are very common in childhood motor disorders, again due to the higher prevalence of cerebral palsy.
Chorea
Chorea describes an apparently random, nonrhythmic, purposeless set of movements of either distal or proximal muscles that appears to flow from one muscle or muscle group to another without any pattern. The causes of chorea and chorealike movements in childhood are summarized in Box 68-1. Chorea occurs at rest and with action, and gives the child a “fidgety” appearance and the inability to remain still. It is associated with motor impersistence (for example, the inability to maintain the tongue extended). Chorea may worsen or improve with voluntary movement, but even very severe chorea may not prevent accurate voluntary movement for some children, suggesting that compensatory mechanisms exist. Many individuals with chorea will incorporate the involuntary movements into a voluntary movement in order to mask the movements. However, the involuntary movements can lead to significant disability, and some children will injure themselves or others due to rapid, ballistic, flinging movements of the arms or legs. Tone is normal or reduced in pure chorea but, in children, chorea may occur in the presence of hypertonic disorders, including dystonia. As with most movement disorders, chorea disappears in sleep but it may be at its worst when the child is drowsy. The term “choreiform” is often used to describe the minimal twitching or “piano-playing” movements seen in many normal young children when arms are extended during the neurological exam. The movements of chorea are briefer than the sustained muscle contractions seen in dystonia and are longer in duration than the “shocklike” movements of myoclonus [Marsden et al., 1983].
Box 68-1 Causes of Chorea in Childhood










































Drugs/Toxins
 Neuroleptic medications, including antiemetics (haloperidol, chlorpromazine, pimozide, prochlorperazine, metoclopramide)
Neuroleptic medications, including antiemetics (haloperidol, chlorpromazine, pimozide, prochlorperazine, metoclopramide)
































Huntington’s Disease
Huntington’s disease (HD) is transmitted as an autosomal-dominant trait, caused by a trinucleotide repeat expansion of the IT-15 gene on chromosome 4 [Li et al., 1993]. This disorder is characterized by a combination of dystonia, chorea, myoclonus, behavioral abnormalities, ataxia, and, ultimately, dementia. When HD begins in childhood, the typical presentation of the movement disorder is dystonia and rigidity, and not chorea. Seizures are not infrequently the first manifestation of HD in childhood. This has been referred to as the Westphal variant of HD [Quinn and Schrag, 1998; Topper et al., 1998]. The age of onset is earlier for children with a higher number of repeats [Langbehn et al., 2004]. There tends to be amplification of the number of repeats, particularly when it is transmitted from father to child, and therefore in most children with juvenile HD disease is inherited from the father and involves repeat lengths that are significantly higher than those seen in adults.
Diagnosis is based upon identification of the trinucleotide repeat sequence. Although at least 38 repeats are usually required for the occurrence of symptoms in adults, a larger number of repeats would be expected when symptoms present in childhood [Quinn and Schrag, 1998; Langbehn et al., 2004]. Magnetic resonance imaging (MRI) shows atrophy of the heads of the caudate nucleus and, in later stages, generalized cerebral and cerebellar atrophy.
In teenagers, the initial presentation of HD may be with psychiatric illness and, in particular, major depression [Tost et al., 2004]. Dystonia or chorea usually supervenes later. The pathology includes basal ganglia, cerebellar, and cortical degeneration [Vonsattel et al., 1985; de la Monte et al., 1988; Myers et al., 1988]. The origin of the chorea has been hypothesized to be a primary loss of medium spiny cells in the striatum that bear D2-like receptors [Deng et al., 2004]. In children, there may be a relatively symmetric loss of D1-bearing and D2-bearing medium spiny neurons, which could account for the primarily dystonic rather than choreic presentation [Augood et al., 1997; Albin et al., 1990].
Treatment of Huntington’s chorea in adults is typically based upon modification of the chorea or myoclonus. Since the chorea has been presumed to be due to loss of medium spiny neurons in the indirect pathway, neuroleptic medications have been most frequently used in adults [Bonelli and Hofmann, 2004; Bonelli et al., 2004]. Neuroleptics would be expected to compensate partially for early loss of indirect pathway neurons by blocking the inhibitory effects of dopamine and disinhibiting the remaining indirect pathway neurons. There is much less experience in children, for whom dystonia and rigidity may be greater contributors to disability. Other medications that have been used successfully in HD include tetrabenazine [Asher and Aminoff, 1981; Ondo et al., 2002], clonazepam [Thompson et al., 1994], and valproic acid [Grove et al., 2000]. There is currently no treatment that modifies the progression of the disease.
Ataxia-Telangiectasia
Despite the name of this disorder, ataxia is not always the presenting finding, and ataxia may be a much less prominent early symptom than upper-extremity chorea. However, the chorea is usually less disabling than the ataxia. Other symptoms associated with ataxia-telangiectasia include dystonia, which is often proximal [Bodensteiner et al., 1980]. Ataxia-telangiectasia (Louis–Bar syndrome) is most commonly due to mutations in the ATM (ataxia-telangiectasia mutated) gene on 11q22–23 [Chun and Gatti, 2004; Coutinho et al., 2004; Gatti et al., 1988]. Mutation in this gene leads to decreased inhibition of cell cycling in the context of injury to nuclear DNA, and this leads to an accumulation of mutations at known “hot spots,” including translocations between regions on chromosome 7 and 14 known to be involved with immune function [Farina et al., 1994]. Therefore, one of the cardinal features of this disorder is a history of frequent sinopulmonary infections [Centerwall and Miller, 1958], which requires very close attention to pulmonary function and aggressive treatment of pulmonary infections at all ages. In later stages, hematologic malignancy may occur [Stankovic et al., 1998]. Children with ataxia-telangiectasia may be particularly vulnerable to injury from ionizing radiation and certain chemotherapy agents, and this must be taken into consideration in planning any treatment for malignancy.
The cause of the cerebellar degeneration is not known [Farina et al., 1994], but the pathology shows a characteristic loss of Purkinje cells, such that, by late stages in the disease, Purkinje cells are often completely absent. The cause of the chorea is not known, and basal ganglia pathology is minimal. Eye movement disorders are well described, and include an inability to suppress, as well as an inability to initiate, saccades [Lewis et al., 1999; Bundey, 1994]. The inability to initiate saccades leads to a characteristic finding of oculomotor apraxia, in which a child will use thrusts of the head in order to position the eyes on the target. Diagnosis is based upon laboratory testing, including decreased immunoglobulin (Ig) G2 and IgA, abnormalities in T-cell subsets, elevated α-fetoprotein, lymphocyte radiosensitivity, and decreased radiation-induced mitotic suppression [Stray-Pedersen et al., 2004]. Treatment of this disorder is primarily supportive, although there are isolated reports of dystonic symptoms responding to trihexyphenidyl or l-DOPA. The chorea generally is mild and does not require specific treatment. There is no known treatment for the ataxia.
Ataxia-oculomotor apraxia is an autosomal-recessive disorder with similar motor symptoms, caused in some cases by mutations in the aprataxin gene AOA-1 [Tranchant et al., 2003; Shimazaki et al., 2002; Moreira et al., 2001]. This disorder commonly presents with chorea, ataxia, and oculomotor apraxia [Sano et al., 2004]. It is most prevalent in Japan and Europe. AOA-1 does not seem to be involved with cell cycle regulation, and therefore the DNA breakage, immune abnormalities, and malignancies seen in ataxia-telangiectasia do not occur. These patients have hypoalbuminemia [Shimazaki et al., 2002]. AOA-2 is a similar autosomal-recessive disorder due to mutations in Senataxin (SETX). Like ATM, Senataxin is involved with DNA repair, although mutations are not associated with increased radiosensitivity [Suraweera et al., 2009]. Clinical features include ataxia, oculomotor apraxia, peripheral neuropathy, and, more rarely, chorea or dystonia [Criscuolo et al., 2006; Le Ber et al., 2004]. The long-term prognosis of AOA-1 and AOA-2 is considerably better than for ataxia-telangiectasia.
Other Genetic Choreas
Other genetic causes of chorea include familial benign hereditary chorea [Kleiner-Fisman et al., 2003; Breedveld et al., 2002a], which is an autosomal-dominant disorder that, in some cases, is due to mutations in the TITF-1 gene on chromosome 14 [Breedveld et al., 2002b]. Affected family members have normal or near-normal intelligence, and chorea is often the only complaint, although mild truncal dystonia and gait ataxia have been reported [Fernandez et al., 2001]. The severity of chorea can vary between different affected family members. It may be accompanied by hypothyroidism, pulmonary disease, or both in some family members with TITF-1 mutations. Symptomatic treatment may yield some mild benefit.
Neuroacanthocytosis is also a potential cause of chorea [Marson et al., 2003], although classification as a primary or secondary chorea is not universally agreed upon.
Sydenham’s Chorea
Of the secondary causes of childhood chorea, Sydenham’s chorea is the most common [Jordan and Singer, 2003; Jummani and Okun, 2001; Garvey and Swedo, 1997]. It has onset weeks or months after an acute infection with group A beta-hemolytic streptococcus (GABHS) and is one of the cardinal symptoms of rheumatic fever. Symptoms may persist for weeks or months, but chorea almost always resolves spontaneously within 6 months [Jordan and Singer, 2003]. There have been rare reported cases of continued or recurring symptoms [Faustino et al., 2003]. The chorea typically involves the distal musculature, initially of one hand and then of both, with a “piano-playing” pattern, but other forms of chorea, including ballism, have been observed. Large, generalized body movements in some patients previously inspired the term “St. Vitus’ dance.”
Anti-basal ganglia antibodies (ABGA) are found in some children [Church et al., 2002] and it has been hypothesized that production of these antibodies is triggered due to molecular mimicry by streptococcal antigens [Loiselle and Singer, 2001; Kirvan et al., 2003; Singer et al., 2003; Goldenberg et al., 1992]. ABGA can be detected in cerebrospinal fluid [Singer et al., 2003], and anti-streptolysin (ASLO) antibodies can be detected in serum. However, the high prevalence of positive ASLO titers in the general population means that both acute and convalescent ASLO titers must be measured in order to probe for an acute infection. The clinical situation often provides a strong indication for the diagnosis, but if other neurological symptoms are present or there is doubt about etiology, a more complete work-up may be needed, including tests for thyroid function, toxins, metabolic disorders, or encephalitis.
Sydenham’s chorea usually does not require treatment, although the acute streptococcal infection should be treated. All children diagnosed with Sydenham’s chorea, even in cases of isolated chorea, should be treated with penicillin, both acutely for treatment and long-term for prophylaxis, according to American Academy of Pediatrics guidelines [American Academy of Pediatrics, 2006].
Penicillin or an acceptable alternative is effective as secondary prevention of recurrent chorea, but more importantly reduces the likelihood that future GABHS infections will cause carditis and permanent valvular damage. Current recommendations in the United States are for prophylactic treatment until age 21 years, regardless of the age of chorea onset. Valproic acid, carbamazepine, or neuroleptics may have some symptomatic benefit in more severe or persistent cases [Pena et al., 2002; Genel et al., 2002; Kulkarni and Anees, 1996]. Treatment with immune suppressant medication, including corticosteroids or intravenous immunoglobulin preparations, has been studied, but the natural history, with spontaneous resolution of symptoms, makes interpretation of efficacy in open clinical trials difficult [Garvey and Swedo, 1997; Green, 1978; Cardoso et al., 2003]. One randomized, blinded, placebo-controlled study showed that a 4-week, 2-mg/kg daily oral dose of prednisone, followed by a taper, reduced duration of chorea and accelerated the reduction in symptoms. Weight gain was substantial by the end of 2 months, and long-term outcome including recurrence rates was not different between groups [Paz et al., 2006]. There are sometimes associated obsessive-compulsive or behavioral symptoms [Moore, 1996; Wilcox and Nasrallah, 1988], and these symptoms are often managed with selective serotonin reuptake inhibitors.
The prognosis for the movement disorder is excellent, and complete resolution occurs in most cases. Recurrence is rare and is sometimes associated with recurrence of other symptoms of rheumatic fever [Korn-Lubetzki et al., 2004]. There appears to be a higher risk of chorea gravidarum in women with a previous history of Sydenham’s chorea [Korn-Lubetzki et al., 2004].
The rapid onset of Sydenham’s chorea has raised the question of whether an autoimmune mechanism could be responsible for other acute-onset movement disorders. This has led to the hypothesis of pediatric autoimmune neuropsychiatric disorders associated with Streptococcus (PANDAS) [March, 2004; Trifiletti and Packard, 1999]. PANDAS is characterized by an abrupt or explosive onset of tics, obsessive-compulsive behavior, and a movement disorder (usually chorea) that is temporally associated with streptococcal infection [Snider and Swedo, 2004; Chmelik et al., 2004; Pavone et al., 2004; Swedo, 2002]. Symptoms may persist, with a relapsing and remitting course. The existence of this disorder has been debated [Singer and Loiselle, 2003] [Swedo et al., 2004; Kurlan and Kaplan, 2004; Kurlan, 2004], and ABGA have not been shown to be elevated [Singer et al., 2004]. It has not been possible to transmit the disorder passively to animals by inoculation with antibodies from affected humans [Loiselle et al., 2004]. Nevertheless, there have been an increasing number of case reports of explosive onset of neurobehavioral disorders, including tic disorders, chorea, and obsessive-compulsive disorder following streptococcal infections, and therefore the hypothesis merits further investigation. It is not clear that there is any specific treatment, although some authors advocate immune modulation, long-term antibiotics, or tonsillectomy [Green, 1978], [Araujo et al., 2002; Heubi and Shott, 2003; van Toorn et al., 2004; Leonard and Swedo, 2001; Gebremariam, 1999].
Medication-Induced Chorea
Chorea is frequently associated with administration of medications, and any child with chorea needs to be carefully examined for the possibility of iatrogenic causes. In particular, treatment of other movement disorders with anticholinergic medication, such as trihexyphenidyl, can precipitate chorea, even at relatively low doses [Nomoto et al., 1987]. Antiepileptic medications, including carbamazepine and phenytoin, have been associated with precipitation of chorea, and in children with diffuse brain injury, sedating medications can sometimes precipitate chorea.
Ballism
Ballism is a high-amplitude, flinging movement, usually due to involuntary movements of proximal joints. Ballism is part of the spectrum of chorea and involves similar pathophysiological mechanisms [Albin et al., 1989; DeLong, 1990]. When ballism involves one side of the body, it is called hemiballism. Hemiballism is the classical manifestation of vascular events affecting the subthalamic nucleus, but can be associated with lesions in other parts of the basal ganglia.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


