Presentations and Diagnostic Tests
Myotonia and Paramyotonia
Myotonia is the phenomenon of delayed muscle relaxation after contraction. It is experienced by patients as stiffness, cramp, or locking of muscles. Stiffness may be mildly worse in the cold or after periods of rest, but diminishes with repeated muscle contractions (the warm-up phenomenon). The clinical findings are an inability to immediately relax muscles after forceful contraction and contraction induced by direct percussion (percussion myotonia). Lid-lag may be seen on rapid downgaze. These findings become less marked with repetition (the warm-up phenomenon) but reappear after rest. In paramyotonia muscle stiffness is precipitated by exercise. This is the opposite of the warm-up phenomenon in myotonia (hence paradoxical myotonia or paramyotonia). Paramyotonia is much more temperature-sensitive than classic myotonia. Exposure to cold not only produces muscle stiffness in paramyotonia, but may also trigger muscle weakness.
 tips and tricks
tips and tricks
- Marked exacerbation by cold and exercise is useful clinically in distinguishing paramyotonia from myotonia. Ask about the effects of eating ice-cream and swimming in cold water.
- Exposure to cold not only produces muscle stiffness in paramyotonia, but also often produces muscle weakness.
Weakness
The hallmark of the periodic paralyses is episodic weakness, which may affect all the limbs, one side, or be extremely focal. Bulbar and respiratory muscles are rarely affected. Attacks of weakness most commonly occur in the morning after waking from sleep. Triggers include stress, cold, fasting (hyperkalemic periodic paralysis or hyperPP), carbohydrate-rich meal (hypokalemic periodic paralysis or hypoPP), and exercise followed by rest. Tendon reflexes are depressed during an attack of periodic paralysis. Some patients with periodic paralysis develop a fixed myopathy, which can be disabling. Mild fixed weakness may develop in patients with myotonia congenita and paramyotonia congenita.
Muscle Hypertrophy
Muscle hypertrophy is characteristic of the nondystrophic myotonias and is a direct result of muscle overactivity. This is different from the pseudohypertrophy seen in some of the dystrophinopathies: the “true” hypertrophy of myotonic disorders results in increased muscle strength. Some individuals are able to participate in sports requiring strength rather than speed or endurance (e.g. Thomsen-type myotonia).
Clinical–Genetic Correlation
Skeletal muscle channelopathies produce a spectrum of clinical phenomena from pure muscle stiffness (myotonia) to pure muscle weakness (periodic paralysis), with overlap syndromes in between. Chloride channel mutations cause myotonia congenita, characterized by classic myotonia, warm-up phenomenon, and no periodic paralysis whatsoever. At the other end of the spectrum, calcium channel and potassium channel mutations both produce pure periodic paralysis without myotonia. Sodium channel disease sits in the central overlapping part of the spectrum and causes four syndromes: pure myotonia, paramyotonia, hyperPP, and hypoPP.
Genetic Testing
The gold standard for the diagnosis of a skeletal muscle channelopathy is identification of the causative mutation from a blood sample. Sequencing ion channel genes is labor intensive and the clinician should suggest which gene to test first based on the clinical manifestations. The genetic laboratory will usually sequence regions of that gene where mutations are commonly found. If a mutation has been identified in another member of the patient’s family, clearly indicating this to the genetics laboratory will enable a more focused search.
 caution!
caution!
A negative genetics test does not necessarily rule out a channelopathy because mutations may occur in regions of channel genes that have not been tested.
Electrodiagnosis
The roles of electrodiagnostic medicine are to indicate the presence of myotonia that is not detectable clinically or the presence of myopathy (e.g. where myotonic dystrophy is a possibility, or in longstanding periodic paralysis), or to exclude other causes of muscle weakness and stiffness (e.g. neuropathy causing cramp).
The long and short exercise tests, in which the compound motor action potential from a convenient muscle (e.g. abductor digiti minimi) is recorded before and after a period of exercise, can give additional information to guide genetic testing.
Muscle Biopsy
Biopsy is generally not usually necessary for the diagnosis of muscle channelopathies. In patients with suspected periodic paralysis and where other diagnostic tests are inconclusive, finding characteristic vacuolar changes or tubular aggregates on muscle biopsy is helpful in establishing the diagnosis.
 science revisited
science revisited
ion channels
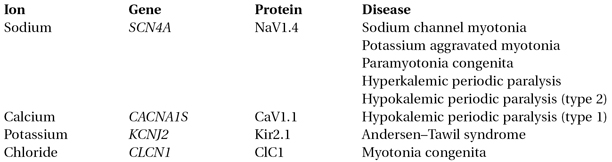
An ion channel is a transmembrane protein that controls the flow of a particular type of ion across a plasma membrane. The channels considered in this chapter are in surface and T-tubule membranes of skeletal muscle cells. An ion channel is formed by the association of several proteins, each protein being identified by a protein name and a gene name, e.g. the SCN4A gene encodes the NaV1.4 subunit of the voltage-gated skeletal muscle sodium channel. Four Nav1.4 proteins associate to form a sodium channel. A single gene can cause different syndromes depending on the mutation (e.g. paramyotonia congenita and periodic paralysis are caused by different mutations of SCN4A). Conversely a syndrome can be caused by mutation of different genes (e.g. myotonia can be caused by CLCN1 or SCN4A mutations). The channels discussed in this chapter are as follows:
The Nondystrophic Myotonias
Myotonia congenita (MC), paramyotonia congenita (PMC), and potassium-aggravated myotonia (PAM) are known as “nondystrophic” to distinguish them from myotonic dystrophy, which is not a primary channelopathy. Myotonic dystrophy causes muscle wasting and weakness, whereas nondystrophic myotonia causes muscle hypertrophy.
Myotonia Congenita – Chloride Channel
MC is caused by mutation of the voltage-gated chloride channel, ClC1, encoded by the CLCN1 gene on chromosome 17. Recessive MC (Becker’s disease) is more common and more severe than the dominant variant (Thomsen’s disease). Muscle stiffness may be slightly worse in the cold, although never to the extent seen in paramyotonia, and improves with exercise. Onset is usually in the second decade and the condition progresses slowly over years. The legs are affected first, giving rise to a disproportionate figure with hypertrophic calf and gluteal muscle, but smaller neck and shoulder girdle muscles. Grip and percussion myotonia and the lid-lag phenomenon are easily elicited. Although later in onset, the recessive form is usually more disabling and may exhibit the following features:
(1) more severe myotonic stiffness
(2) transient weakness that accompanies the myotonic stiffness
(3) minor distal wasting and weakness.




