CHAPTER 85 MUSCULAR DYSTROPHIES
Muscular dystrophies constitute a clinically and genetically heterogeneous group of skeletal muscle-wasting diseases.1 The molecular causes of the muscular dystrophies remained elusive for many decades, and the muscular dystrophies were classified into relatively few clinical entities. In the late 1980s, major advances in molecular genetics led to the discovery of the dystrophin gene and its protein product, dystrophin.2,3 Mutations in the dystrophin gene result in dystrophin deficiency, which is the pathogenic determinant of the dystrophinopathies.4,5 Several muscular dystrophies are caused by mutations in other genes that cause defects of proteins localized at the sarcolemma, the cytoplasm or the nuclear envelope. Since the mid-1990s, an increasing number of genes have been associated with different forms of muscular dystrophy (Table 85-1, Fig. 85-1). These findings have led to a profound change in the classification of muscular dystrophies, with a new emphasis on molecular genetics rather than on clinical symptoms (which differentiated diseases on the basis of age at onset, severity, mode of inheritance, and muscle groups primarily affected).


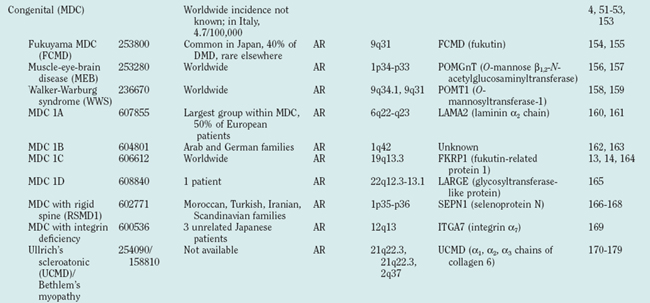
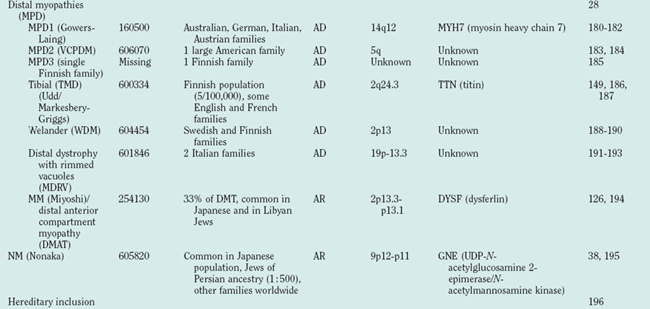

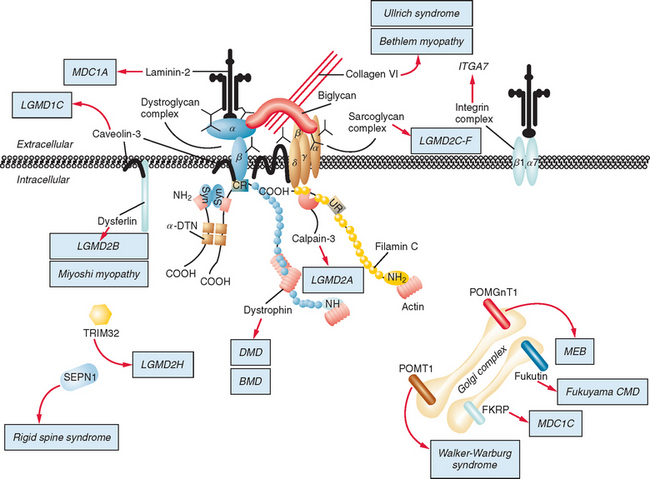
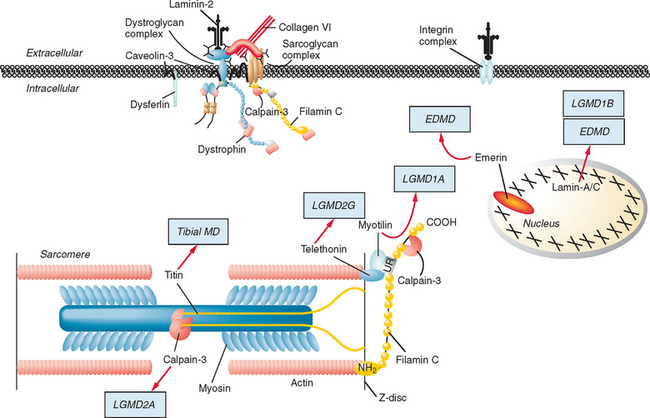
Figure 85-1 Muscular dystrophies and the membrane and enzymatic proteins they are associated with. This schematic shows the locations of various membrane and enzymatic proteins associated with muscular dystrophies. The diseases these molecules cause when mutated are shown in boxes. Dystrophin, through its interaction with the dystroglycan complex, connects the actin cytoskeleton to the extracellular matrix. Intracellularly, it interacts with dystrobrevin (α-DTN) and syntrophins (Syn) (shown in blue). Extracellularly, the sarcoglycan complex (orange) interacts with biglycan, which connects this complex to the dystroglycan complex and the extracellular matrix collagen. Intracellularly, δ- and γ-sarcoglycans interact with filamin C. The majority of filamin C is at the Z-disc of the sarcolemma shown in Figure 85-2. The four proteins shown in the Golgi complex have been demonstrated to affect the glycosylation state of the α-dystroglycan and mediate its binding to the extracellular matrix. Fukutin and fukutin-related protein have been shown to localize to the medial Golgi complex. The localization of POMT1 and POMGnT1 is unknown so far, but the authors hypothesize that they are in the Golgi complex, because they are involved in the glycosylation process. BMD, Becker muscular dystrophy; CMD and MDC, congenital muscular dystrophy; DMD, Duchenne muscular dystrophy; LGMD, limb girdle muscular dystrophy; MEB, muscle-eye-brain disease.
(Reprinted from Dalkilic I, Kunkel LM: Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev 2003; 13:231-238.)
The most common form is the X-linked recessive Duchenne muscular dystrophy (DMD), named after Guillaume Benjamin Amand Duchenne, who described it in 1861.6,7 Positional cloning of the gene altered in DMD8 led to the discovery of dystrophin2 and to improved molecular diagnosis of DMD and of its milder allelic variant, Becker muscular dystrophy (BMD).9
About 50 years after Duchenne, Batten10 published the first cases of congenital muscular dystrophy (MDC). Unlike patients with the DMD/BMD phenotype, patients with MDC present with weakness and dystrophic changes in the muscle biopsy at birth, but symptoms are less rapidly progressive than in DMD.
The term limb girdle muscular dystrophy (LGMD) was introduced in the middle of the 20th century, when it became obvious that there was an additional major group of noncongenital muscular dystrophies that differed from both the X-linked DMD and BMD and from the autosomal-dominant facioscapulohumeral forms.11 Nowadays, the term limb girdle muscular dystrophy has changed from a wastebasket designation to an ever-expanding list of subtypes (18 thus far), for which accurate molecular diagnoses are available. Age at onset usually ranges from early childhood to late adulthood12; however, the same gene defect can cause allelic forms of MDCs and LGMDs, as shown for the fukutin-related protein deficiencies.13–15
The finding that mutations in nuclear envelope proteins also cause muscular dystrophies—referred to as nuclear envelopathies or Emery-Dreifuss muscular dystrophies—came as a surprise when emerin, the protein shown to be mutated in X-linked Emery Dreifuss muscular dystrophy (EDMD1, X-EDMD)16 was also found to be an inner nuclear membrane protein (Fig. 85-2). The importance of the nuclear envelope in neuromuscular disease was bolstered by the discoveries that mutations in the LMNA gene can cause autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD2),17 dilated cardiomyopathy with conduction defect (CMD1A),18 limb-girdle muscular dystrophy type 1B (LGMD1B),19 Charcot-Marie-Tooth disorder type 2B1 (CMT2B1),20 and a variety of other, nonneuromuscular diseases, such as Dunnigan’s familial partial lipodystrophy and mandibuloacral dysplasia, or premature aging syndromes, such as Hutchinson-Gilford progeria and atypical Werner’s syndrome.21,22 A new phenotype combining myopathy and progeria has been described.23
Facioscapulohumeral muscular dystrophy (FSHD), first described in 1885,24 is a frequent form of muscular dystrophy that has a distinctive clinical manifestation with autosomal dominant inheritance.11 The gene underlying FSHD was mapped to chromosome 4q35 in 199225 and was shown to be closely linked to locus D4F104S1. Although D4F104S1-associated deletions are closely associated with FSHD, the identity and location of the FSHD gene (or genes) still remain elusive, as does the mechanistic basis of the disease.
Oculopharyngeal muscular dystrophy (OPMD) is unusual among muscular dystrophies because of its manifestation in late adult life, typically in the sixth decade. Symptoms include progressive ptosis and dysphagia, followed by involvement of other cranial and limb muscles. OPMD is usually inherited as an autosomal dominant trait as a result of an expanded guanine-cytosine-guanine (GCG) repeat detectable in the poly A binding protein 2 gene on chromosome 14.26,27
Distal myopathies are frequent in the Nordic countries and are being increasingly recognized elsewhere. To date, six different distal myopathy phenotypes have been identified, and there has been considerable progress in the understanding of the molecular pathophysiology underlying the distal myopathies.28 Mutations in membrane-associated dysferlin cause two different distal phenotypes, allelic to LGMD2B,29 whereas mutations in titin can cause either a distal myopathy (type Udd/Markesbery-Griggs) or LGMD2J,30,31 another example of the diversity between clinical and genetic disease definitions.
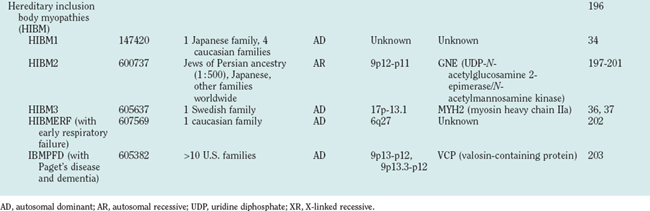
Hereditary inclusion body myopathies (HIBM) belong to the heterogeneous group of myopathies with rimmed vacuoles. So far, autosomal recessive (HIBM2)32 and autosomal dominant forms33–37 have been linked to different gene loci. Interestingly, distal myopathy with rimmed vacuoles (Nonaka myopathy) has been attributed to mutations in the GNE gene and is therefore allelic to HIBM2.38
DYSTROPHINOPATHIES
Clinical Features
In DMD, children usually come to medical attention between ages 3 and 5 years because of frequent falls, awkward running, and waddling gait. At this stage, weakness is most pronounced in the hips and proximal leg muscles but over time will affect neck flexors, proximal arm muscles (especially biceps), ankle dorsiflexors, and respiratory muscles, necessitating ventilation at some point during the course of the disease. Calf hypertrophy is a distinctive feature, whereas facial and bulbar muscles are largely spared. Creatine kinase levels are massively elevated, 50 to 100 times normal. The disease progresses relentlessly, and ambulation is lost between 7 and 12 years of age. Prednisone, the primary pharmacological therapy for DMD, may modestly prolong ambulation.
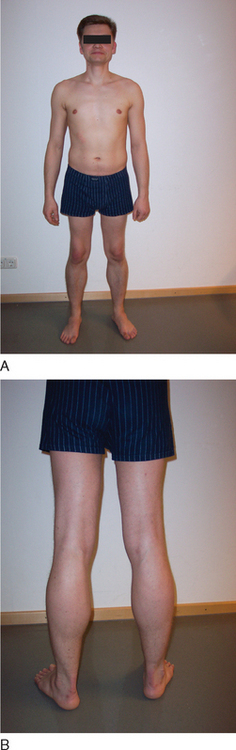
BMD is an allelic variant of DMD and has a more benign and variable manifestation with later onset and slower progression. Onset of symptoms after age 6 or walking after age 13 are typical features. In rare cases, BMD is not diagnosed until adulthood and the patients never lose ambulation (Fig. 85-3).
Female carriers of DMD or BMD are usually asymptomatic but may also have limb weakness, elevated creatine kinase levels, and nonmuscular symptoms such as cardiac involvement. The extent of clinical severity depends in part on the degree of skewed X chromosome inactivation in somatic cells.39
Skeletal muscle weakness in DMD and BMD is frequently accompanied by cardiac muscle dysfunction. Approximately 95% of patients have cardiac involvement by the time of death. This includes electrocardiographic abnormalities, arrhythmias, and myocardial dilatation and thickening. Patients with DMD and BMD may have normal intelligence, but the mean Wechsler full IQ is below average: 25% of BMD and 31% of DMD patients have an IQ of less than 75. Scoliosis is frequently severe. Contractures develop in hips, ankles, and elbows. On occasion, involvement of smooth muscle leads to gastrointestinal complications, including pseudo-obstruction.40
Etiology and Pathophysiology
Dystrophin is the largest human gene, covering 2.5 megabases and including 79 exons. The enormity of the gene, along with the spontaneous mutation rate of each base pair allows a high frequency of novel mutations, which explains how 30% of all DMD and BMD cases result from spontaneous mutations. The gene encodes a 427-kD subsarcolemmal protein. Dystrophin has several well-characterized domains, including an amino-terminal domain with homology to α-actinin, a large “rod” domain with spectrin-like repeats, a cysteine-rich domain, and a carboxy-terminal domain with homology to other dystrophin-related proteins. Dystrophin interacts with and stabilizes a large membrane-associated protein complex, the dystroglycan complex, whose components are involved in other muscular dystrophies (see Fig. 85-1). It links the actin cytoskeleton through its amino-terminal binding domain to the sarcolemma and extracellular matrix through interactions with its carboxy-terminal binding domains. In addition to physically connecting and stabilizing this large protein network, dystrophin may play a role in signal transduction because of its association with nitric oxide synthase and highly phosphorylated postsynaptic proteins.
Approximately two thirds of disease-causing mutations in dystrophin are large deletions; an additional 5% are duplications within hot spots of the dystrophin gene. These common mutations can be detected by commercially available multiplex polymerase chain reaction, which amplifies 18 to 25 of the gene’s 79 exons from genomic DNA obtained from blood samples. Additional deletions and duplications may be detected by amplifying and quantifying all exons. There is no simple relation between size of the deletion and resultant clinical phenotype. For example, deletion of small exons, such as exon 44, often results in classic DMD, whereas large deletions that may involve nearly 50% of the gene have been described in patients with BMD. The central and distal rod domains seem to be functionally almost dispensable, inasmuch as some deletions in this region are associated only with myalgia or muscle cramps, not with weakness. Some patients may even present solely with an isolated increase in creatine kinase level. This has been shown in patients with in-frame deletions in exons 32 to 44, 48 to 51, or 48 to 53, all of whom had normal or near-normal dystrophin levels at the sarcolemma. The effects on the phenotype depend, therefore, not so much on the size of a deletion (or duplication) but on whether it disrupts the reading frame. A further observation is that deletions very different in size and position may produce very similar severe phenotypes. The reason for this effect might be the occurrence of nonsense-mediated RNA decay. This phenomenon may account for the lack of rescue of dystrophin function, as well as for the phenotypic variability related to variations in the efficiency of RNA decay control. Mutations that maintain the reading frame (in-frame mutation) generally result in abnormal but partially functional dystrophin and are associated with BMD. In patients with DMD, deletions and duplications disrupt the reading frame (frame-shift mutations), resulting in unstable RNA and nearly undetectable truncated proteins. The reading frame hypothesis holds true for over 90% of cases and is commonly used both as a diagnostic confirmation of dystrophinopathies and for the differential diagnosis of DMD and BMD.41
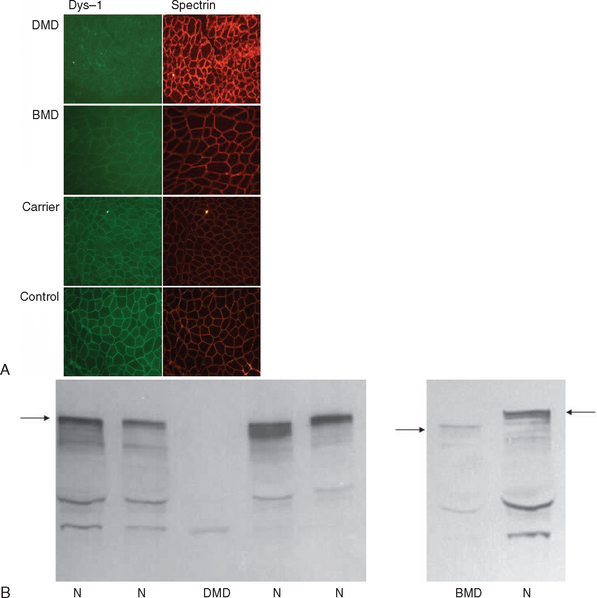
If knowledge of a specific small mutation is desired, complementary DNA may be generated from muscle messenger RNA and sequenced. Single-strand conformation polymorphism analysis42 and single-condition amplification/internal primer sequencing43 of genomic DNA have also been proposed for identification of small mutations. Furthermore, Buzin and associates reported mutation rates in the dystrophin gene with a mutational hot spot at a cytosine-guanine island (CpG) dinucleotide.44 Approximately 30% are point mutations and 10% to 15% are nonsense mutations that produce premature stop codons. If genomic DNA testing does not reveal a deletion or duplication, a muscle biopsy is frequently performed to confirm the diagnosis. Histological features include marked variability of fiber size with aberrant large fibers and hypercontracted muscle fibers along with groups of atrophic, degenerating, and regenerating fibers. Endomysial fibrosis and fatty infiltration are invariably present along with some mononuclear inflammatory cell infiltration. Dystrophin immunostaining shows absence of the protein in DMD and patchy staining in BMD and may reveal a mosaic pattern of expression in female carriers. Dystrophin immunoblotting shows absence of the dystrophin band in DMD, shows weak expression and/or abnormal molecular weight in BMD, and usually yields normal results in female carriers (Fig. 85-4).
LIMB GIRDLE MUSCULAR DYSTROPHIES
Definition
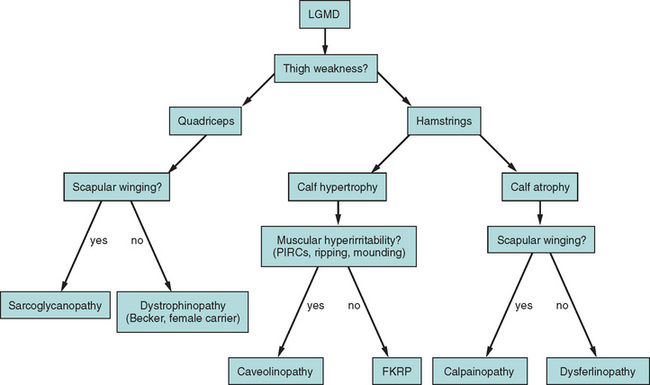
Because of molecular discoveries since the mid-1990s, LGMD has emerged as an entire field within the inherited myopathies; at least six autosomal dominant and ten autosomal recessive gene defects have been identified. Redefined in 1995, the LGMD are face-sparing, predominantly proximal, progressive muscular dystrophies with elevated creatine kinase levels, and dystrophic features are demonstrated on muscle biopsy (Fig. 85-5).12,45–47 In the current classification system, LGMDs are divided into autosomal dominant (LGMD1) and autosomal recessive (LGMD2) disorders with an additional lettering system that denotes the chronological order of chromosomal linkage (thus far, A through G for autosomal dominant LGMD and A through K for autosomal recessive LGMD). Accurate diagnosis is now possible for most LGMDs and is important for genetic counseling. Certain LGMDs are characterized by treatable cardiac and respiratory complications.45,48
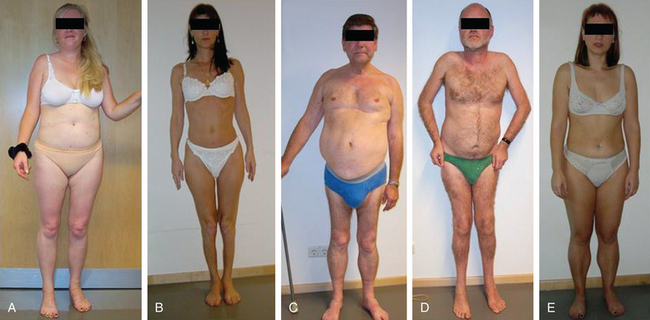
The causes of the LGMD include mutations in a wide range of proteins and protein systems. Calpain, mutated in LGMD2A, is a calcium-activated neutral protease, and this is the first known genetic defect in muscular dystrophy stemming from an enzyme defect. Other mutations impair membrane signaling and repair (caveolin, dysferlin), sarcolemmal integrity (sarcoglycans), and contractile dysfunction (myotilin, telethonin, titin). The subcellular localization of these proteins also varies from the nuclear membrane (lamin A/C) to the sarcomere (myotilin, calpain, telethonin, titin), the sarcoplasm (TRIM32, a putative E3-ubiquitin-ligase), the sarcolemma (caveolin, sarcoglycans, dysferlin), and even the extracellular space (fukutin-related protein). Of note, involvement of one nuclear membrane protein, emerin, causes X-linked Emery Dreifuss syndrome (X-EDMD, EDMD1), whereas mutations in the related protein, lamin A/C, cause EDMD2. It remains unclear how mutations in apparently unrelated proteins can cause similar phenotypes, and it is difficult at present to see a common final pathway of action. Likewise, identical genotypes often yield substantially different phenotypes even within the same family. Dysferlinopathies can cause LGMD2B or distal myopathies, and caveolinopathies have been associated with LGMD1C, rippling muscle disease, persistent elevated creatine kinase levels (hyperCKemia), or distal myopathy. Fukutin-related protein gene (FKRP) mutations manifest high clinical variability, with onset at birth or later in adulthood (LGMD2I) (Fig. 85-6). Titinopathies exhibit phenotypic variability depending on the amount of genes involved: A single mutated allele causes a late adult-onset distal myopathy (Finnish tibial muscular dystrophy), whereas two abnormal alleles cause LGMD2J. This remarkable phenotypic variability remains enigmatic, but explanations may be forthcoming through investigations of modifier genes, DNA microarrays, and proteomic studies.49 LGMD2K has been described as an allelic form of Walker-Warburg syndrome in Turkish patients.50 Further phenotypic differentiation of the LGMDs is provided in Table 85-2 and Figure 85-7.

Autosomal Dominant Limb Girdle Muscular Dystrophy
Autosomal dominant LGMDs tend to have an altogether slower course and later onset with less elevation of serum creatine kinase level in comparison with autosomal recessive LGMDs. They are also clinically much more heterogeneous (see Table 85-2 and Fig. 85-7). Except for LGMD1C (caveolinopathy), immunohistochemistry and immunoblotting are of little help in differentiating dominant LGMD subtypes.
Autosomal Recessive Limb Girdle Muscular Dystrophy
The autosomal recessive forms of LGMD for which the genetic bases are known can be further subdivided on the basis of the genes and proteins involved (see Table 85-1). The first autosomal recessive form, LGMD2A, was mapped to chromosome 15q in 1991 in patients from the Reunion Island. Ten additional forms have been mapped since the mid-1990s, most recently LGMD2K. The protein products of these 11 genes have been identified (see Fig. 85-1): calpain-3 for LGMD2A, dysferlin for LGMD2B, γ-sarcoglycan for LGMD2C, α-sarcoglycan for LGMD2D, β-sarcoglycan for LGMD2E, δ-sarcoglycan for LGMD2F, the sarcomeric protein telethonin for LGMD2G, TRIM32 for LGMD2H, fukutin-related protein for LGMD2I, titin for LGMD2J, and POMT1 for LGMD2K. Genotypephenotype correlation studies have been reported for the different forms of LGMD in an attempt to enhance comprehension of the underlying pathological mechanisms, to better characterize each of the subgroups and, of more importance, to identify modifier genes or epigenetic factors that might modulate the clinical course in patients who carry the same pathological mutation. Immunohistochemistry and immunoblotting are pivotal for LGMD2 subtype differentiation and for directing the molecular genetic analysis (see Table 85-2 and Fig. 85-7).
CONGENITAL MUSCULAR DYSTROPHIES
Definition
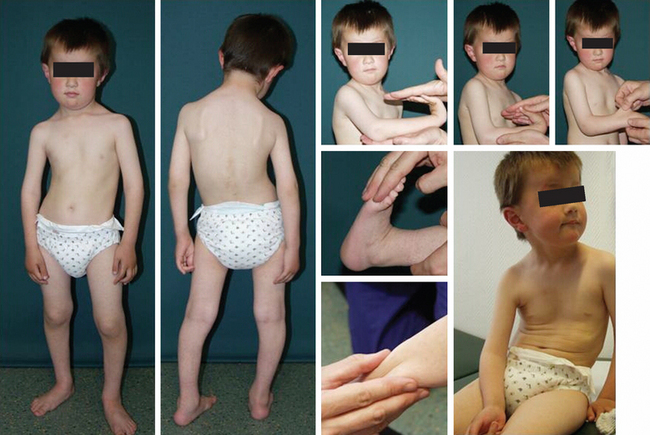
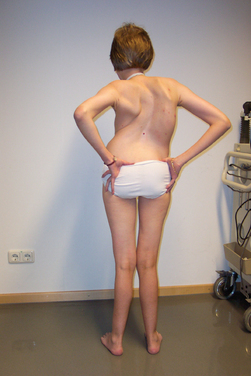
The MDCs are a group of genetic disorders in which weakness and abnormal muscle histological features are present at birth. Muscle weakness tends to be more stable, but complications can become more prominent over time. A firm diagnosis of MDC requires a muscle biopsy. Pathological findings include variation in fiber size, internal nuclei, and increased endomysial and fatty tissue. Signs of ongoing degeneration and regeneration may be less prominent than in muscular dystrophies of later onset (e.g., DMD, BMD, or sarcoglycanopathies). Creatine kinase concentrations are variable and can be normal. The mode of inheritance for most MDCs is autosomal recessive with significant genetic heterogeneity.51–53 For clinical differentiation, see Table 85-2 and Figs. 85-8 and 85-9.
Clinical Features, Etiology, and Pathophysiology
Immunohistochemistry and immunoblotting help to distinguish between merosin-positive and merosin-negative forms of MDC. Reduction or absence of α-dystroglycan immunostaining indicates a defect of one of the glycosylation genes (see Tables 85-1 and 85-2 and Fig. 85-6).
NUCLEAR ENVELOPATHIES
Definition
Emery Dreifuss muscular dystrophies are characterized by early contractures of the elbows, Achilles tendons, and spine; slowly progressive muscle wasting and weakness with a predominantly humeroperoneal distribution; and cardiomyopathy, usually manifesting as heart block. There are two main modes of inheritance: X-linked (X-EDMD, EDMD1) and autosomal dominant (ADEDMD, EDMD2/LGMD1B). However, a rare autosomal recessive mode of inheritance (EDMD3) has also been reported.54 In 1999, the nuclear lamin A/C gene, LMNA, at locus 1q11-q23 was found to be responsible for EDMD2 and EDMD3.17 Whereas mutations in the emerin gene, STA, at locus Xq28, give rise to one phenotype, the X-linked form of Emery Dreifuss muscular dystrophies, mutations in LMNA cause at least nine (and probably more) different neuromuscular and nonneuromuscular phenotypes.55 For clinical differentiation, see Table 85-2.
Etiology and Pathophysiology
One of the most intriguing questions regarding the role of emerin and lamins A and C in disease is how mutations in nearly ubiquitously expressed proteins can cause different tissue-specific illnesses. Several hypothetical pathophysiological mechanisms have been proposed. Protein complexes of the nuclear envelope, which include lamins and emerin, may function in the regulation of gene expression, inasmuch as there are several examples of nuclear envelope proteins that interact with chromatin proteins. The functional effects of these interactions are not yet understood, but they suggest a potential role for the nuclear envelope in chromatin organization. Therefore, alterations in the nuclear envelope that result from mutations in emerin and lamins A and C may change the expression of genes involved in striated muscle function. Another hypothesis is that mutations in nuclear envelope proteins cause neuromuscular disease by altering the structural integrity of the nucleus. This so-called “mechanical stress” hypothesis could partly explain the muscle-specific phenotypes of some of these diseases. Nuclei with mutant structural proteins may be more fragile. During muscle contraction, nuclei in muscle cells may be under more stress than are nuclei of other cells and therefore may be sensitive to defects in the nucleoskeleton.56
FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
Definition
FSHD is a highly variable disorder in which weakness appears from infancy to late life but typically in the second decade. Usually, the disease initially involves the face and scapulae, followed by foot dorsiflexors and hip girdles. Typical features include the often striking asymmetry of muscle involvement and the sparing of bulbar extraocular and respiratory muscles.57
Clinical Features
Clinical diagnosis rests on the distinctive pattern of muscle involvement (Fig. 85-10). Although several inherited myopathies can include facial and shoulder girdle weakness, the following features are specific to FSHD: (1) Facial weakness is prominent and typically more severe in the lower facial musculature. (2) Extraocular, eyelid, and bulbar muscles are spared. (3) The shoulders anteriorly have a typical contour with straight clavicles, forward sloping, and rounding. (4) Pectoral, biceps, and triceps muscles are typically involved, whereas deltoid and forearm muscles are spared. (5) Lower abdominal muscles are selectively involved, resulting in lumbar lordosis. (6) Leg involvement frequently starts with foot dorsiflexor weakness, but quadriceps weakness can also be manifest early in the disease.57 (7) Contractures are rarely present, but during the course of the disease, patients may develop additional weakness of wrist extensors. (8) Muscle weakness is often asymmetrical.
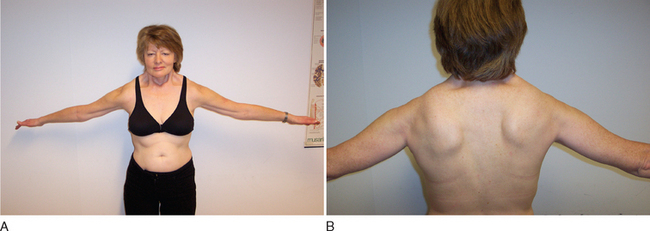
Extramuscular manifestations of FSHD include high-frequency hearing loss, retinal telangiectasias, and a tendency to develop atrial arrhythmias.58 These manifestations, if present, are mostly mild. Symptomatic cardiac involvement, present in approximately 5% of patients with molecularly confirmed FSHD, consists of conduction abnormalities with frequent supraventricular tachyarrhythmias.
Inheritance is autosomal dominant with a high degree of penetrance, but sporadic cases caused by de novo mutations account for up to 30% of cases.59
Etiology and Pathophysiology
Understanding the pathophysiology of FSHD is hampered by the unique molecular genetic lesion. Despite the identification of a causal deletion on chromosome 4q35 in 1990, the molecular pathophysiology of FSHD remains unclear. When FSHD was linked to chromosome 4q35, subtelomeric rearrangements consisting of deletions of an integral number of copies of a 3.3kb DNA repeat named D4Z4 were identified.25 Whereas normal individuals have 15 or more repeats on each copy of 4q35, individuals with FSHD have 12 or fewer repeats on each copy. The deletions do not appear do disrupt an expressed gene. Rare FSHD kindreds fail to show linkage to 4q35, but alternative genetic loci have not been identified thus far.
Whereas it is clear that a critical deletion within the 4q35 D4Z4 repeats results in FSHD, no expressed sequences are present within the deleted segments. Investigators have found 4q35 genes located upstream of D4Z4 to be inappropriately overexpressed in FSHD muscle. An element within D4Z4 has been shown to behave as a silencer that provides a binding site for a transcriptional repressing complex. These results are suggestive of a model in which deletion of D4Z4 leads to inappropriate transcriptional repression of 4q35 genes, resulting in disease.60 FRG2, an FSHD candidate gene, has been found to be transcriptionally upregulated in differentiating primary myoblast cultures from patients with FSHD and is an attractive candidate culprit.61 In most cases, diagnosis can be achieved by blood genetic testing without a prior muscle biopsy.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


