science revisited
- Of those with MG 85% present with ocular symptoms.
- The likelihood of progression to generalized MG is low if a patient does not generalize within 2 years of developing ocular symptoms.
Bulbar symptoms are the presenting feature in about 15% of cases. Dysarthria is described as breathy, nasal speech due to palatal weakness, in contrast to the spastic dysarthria of amyotrophic lateral sclerosis (ALS). Patients report liquids escaping through the nose when drinking due to palatal weakness and fatigue with chewing. Weakness of the orbicularis oris muscle causes the classic myasthenic snarl caused by the inability to raise the corners of the mouth when attempting to smile.
Neck flexion is typically more affected than neck extension but rare patients can present with a dropped head syndrome. Proximal limb weakness is usually symmetric. Respiratory weakness is not uncommon in severe generalized MG. Myasthenic crisis is due to severe respiratory weakness and/or severe dysphagia and can be potentially fatal without mechanical ventilation and intensive care support.
Pathophysiology
The weakness observed in MG is due to alteration of neuromuscular transmission at the neuromuscular junction, and reduced ability to depolarize muscle caused by elevated serum antibodies against the postsynaptic ACh receptors (seropositive MG). A subset of patients without ACh receptor antibodies has elevated titers of muscle-specific kinase (MuSK) antibodies. MuSK is believed to help cluster ACh receptors at the postsynaptic neuromuscular junction. The development of ACh receptor antibodies is both T- and B-cell dependent. Alteration in neuromuscular transmission in MG is multifactorial and ultimately leads to loss of the normal folded pattern of the postsynaptic membrane.
Thymic pathology is often observed in MG; 65% of individuals with MG have thymic hyperplasia and 10% have a thymoma. Patients with thymoma typically have more severe phenotypes of generalized MG. Most thymomas are encapsulated and amenable to complete resection; occasional highly invasive malignant thymomas are observed.
Diagnosis
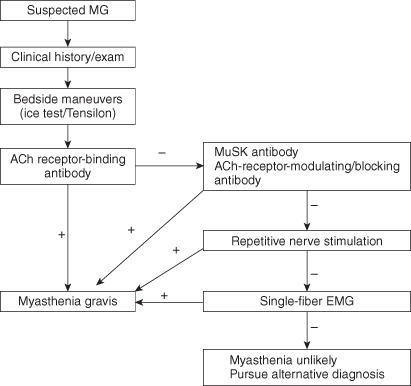
The diagnosis of MG requires a combination of clinical history, physical examination, and confirmatory tests. The authors recommend a stepwise approach to diagnosis (Figure 16.1). The differential diagnosis of MG is short (Box 16.1).
Box 16.1. Differential diagnosis of myasthenia gravis
Neuromuscular junction disease
Lambert–Eaton myasthenic syndrome
Botulism
Tick paralysis
Anterior horn cell disease
Amyotrophic lateral sclerosis
Peripheral nerve
Acute inflammatory demyelinating polyneuropathy
Chronic inflammatory demyelinating polyneuropathy
Myopathy
Ocular pharyngeal muscular dystrophy
Progressive external ophthalmoplegia
History and Bedside Examination
Obtaining a quality clinical history in suspected MG is imperative because the degree of MG weakness fluctuates; an affected patient may have a normal bedside exam in early or partially treated disease. All patients should be asked if they are experiencing the following: diplopia, blurred vision, chewing fatigue, choking, loss of liquids from nose, dysarthria, shortness of breath, and difficulty with repetitive proximal limb tasks (e.g. brushing hair, standing from a chair).
Ice Test
The application of an ice bag to the eyelids for 2–5 min can partially or fully resolve ptosis but does not impact other signs of MG, including dysconjugate gaze. The sensitivity ranges from 84% to 92% and specificity from 97% to 98%.
Edrophonium Chloride Test
Edrophonium chloride (Tensilon) is a short-acting acetylcholinesterase inhibitor that can be administered at the bedside. The medication achieves effect in about 30 s and its duration of action is about 10 min. An initial test dose of 2 mg is injected intravenously. If the patient tolerates the dose, an additional injection of 8 mg is given. A positive result is defined as unequivocal improvement in strength in an involved muscle in 2–5 min. The sensitivity of the test ranges from 60% to 95% in ocular MG and from 71% to 95% in generalized MG.
 caution!
caution!
Atropine (1–2 mg) should be available during a Tensilon test in case the patient develops the rare complication of severe bradycardia and hypotension.
Laboratory Testing
ACh-Receptor Antibodies
Approximately 70–80% of patients with generalized MG have positive antibodies directed against the ACh receptor. Only about 50% of ocular MG patients have positive ACh-receptor antibodies. The presence of ACh-receptor antibodies is reported to be about 97–98% specific for MG. Occasional false-positive results are reported in patients with asymptomatic thymoma and other autoimmune diseases. In patients with typical symptoms of ocular or generalized MG, positive ACh-receptor antibodies should be considered diagnostic.
 tips and tricks
tips and tricks
- ACh-receptor antibodies are 97–98% specific for MG.
- Positive ACh-receptor antibodies are diagnostic in patients with typical symptoms of MG.
MuSK Antibodies
Approximately a third of patients with seronegative MG have antibodies directed against MuSK. This is approximately 7% of the total cases of MG and raises the probability of detectable antibodies in generalized MG to 87–90%. Most MuSK-positive patients are females, and MuSK antibodies should be checked only in cases where ACh-receptor antibodies are negative.
Seronegative MG
Antibodies are not detected in 10–13% of generalized MG cases and diagnosis must be confirmed by electrodiagnostic testing.
 tips and tricks
tips and tricks
- 10–13% of generalized MG cases have no detectable antibodies.
Electrodiagnostic Evaluation
Routine electromyographic (EMG)/nerve conduction studies are useful to exclude non-neuromuscular junction causes of weakness (e.g. ALS, myopathy, chronic inflammatory demyelinating polyneuropathy [CIDP]) and presynaptic neuromuscular junction disease (e.g. Lambert–Eaton myasthenic syndrome and botulism).
Slow Repetitive Motor Nerve Stimulation
During repetitive motor nerve stimulation (RNS), a motor nerve is stimulated at 2–5 Hz in trains of six stimulations at rest and then after a period of exercise. This results in depletion of ACh stores at the neuromuscular junction and reduces successful competition of ACh for the limited ACh receptors in MG. It is imperative to test clinically affected muscles to improve the likelihood of a positive result. The sensitivity of RNS ranges from 53% to 100% in generalized MG and 10% to 20% in ocular MG.
Single-Fiber EMG
Single-fiber EMG (SFEMG) is the most sensitive test to detect pathology at the neuromuscular junction. Increased variability in the time two muscle fibers of the same motor unit depolarize in relation to each other is observed in MG (jitter).
When facial and limb muscles are examined, SFEMG is reported to be 97% sensitive. A normal SFEMG of a weak muscle performed by an experienced electromyographer essentially excludes the diagnosis of MG.
 tips and tricks
tips and tricks
SFEMG has high sensitivity but low specificity for MG, and must be combined with a clinical history and physical exam to establish a diagnosis.




