Myelomeningocele
Myelomeningocele (MMC) represents a primary failure of neurulation and results in an exposed segment of spinal cord on the back of an infant; MMC is the most severe central nervous system (CNS) malformation compatible with life. Improved nutrition and periconceptional folate supplementation have significantly reduced the incidence of this disorder. Improved treatments, including valved shunt systems for hydrocephalus, surgical treatments for symptomatic Chiari malformations and spinal cord tethering, clean intermittent catheterization (CIC) of the neurogenic bladder, and orthopedic surgical procedures and external orthoses to improve ambulation and mobility, have greatly improved the outlook for these children; many are entering adulthood, and transitional issues have consequently assumed significance. This chapter reviews various facets of MMC in childhood.
24.1 Embryology and Genetics of Myelomeningocele
Open neural tube defects (NTDs), such as MMC and anencephaly, are primarily caused by a localized failure of neurulation,1 although a few may arise from overdistention of a previously closed neural tube.2,3 NTDs in animals have been produced by teratogens (reviewed by Campbell et al4), genetic mutations (reviewed by Copp et al5,6), and experimental manipulations (reviewed by Schoenwolf and Smith7), although the cause of human malformations remains unknown. NTDs are likely etiologically heterogeneous4–6,8 and represent the end result of a variety of embryonic disorders.
The importance of genetic factors is supported by the increased prevalence of NTDs among certain ethnic groups,9–11 in association with certain genetic syndromes12–15 and chromosomal defects,15–19 and among families with NTDs.9–11,20 Over 200 genetic mouse models with NTDs have been identified.21,22 Candidate genes are involved in the retention and metabolism of folate23–28 and vitamin B12,29 the methylation cycle and transulfuration,26,28,30,31 glucose transport and metabolism,32 oxidative stress,33 retinoid metabolism, transcription factors, and DNA repair. Folate (vitamin B6) is particularly important. Periconceptional folate reduces both the incidence of NTD among women who have not previously had an infant with NTD (400 µg/d) and recurrences among those with a previously affected pregnancy (4,000 µg/d).34,35 Folate and its metabolites are involved in purine and pyrimidine synthesis and the transfer of methyl groups during the metabolism of methionine and homocysteine (▶ Fig. 24.1). Maternal or fetal mutations of enzymes involved in this “methylation cycle” (▶ Fig. 24.1) may produce methionine deficiency and/or homocysteine excess that can be overcome with supplemental dietary folate.36–38 Teratogens like valproic acid disrupt these metabolic pathways.38,39 Folate-dependent developmental regulatory genes coding for transcription and cell cycle checkpoint factors may also be involved.
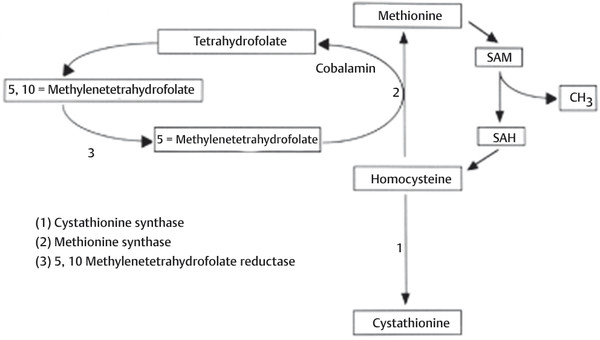
Fig. 24.1 Homocysteine/methionine cycle. The conversion of homocysteine to methionine is mediated by the donation of a methyl group from 5-methyltetrahydrofolate; in the process, tetrahydrofolate is formed. This reaction is catalyzed by the enzyme methionine synthase; cobalamin (vitamin B12) is used as a coenzyme. 5-Methyltetrahydrofolate is regenerated from tetrahydrofolate with 5,10-methylenetetrahydrofolate used as an intermediary, completing a “folate cycle. ” Methionine is subsequently used as a “methyl (CH3) donor” in a variety of important metabolic reactions and is converted to homocysteine in the process. SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine.
The Chiari 2 malformation is likely an embryologically “acquired” malformation. One theory has proposed that venting of fluid during early embryonic development causes the formation of a smaller posterior fossa from mesenchyme; subsequent growth of the cerebellum within a small posterior fossa leads to downward and upward displacement of the cerebellum and brainstem, “beaking” of the midbrain, and enlargement of the tentorial notch, as well as disordered neuronal histogenesis resulting in callosal dysgenesis, cortical heterotopias, and stenogyria (see box “Chiari-Associated Central Nervous System Malformations”).40–42 The posterior fossa herniation may be reversible following fetal MMC closure at up to 28 weeks of gestation, although the associated cerebral malformations persist.43
Chiari-Associated Central Nervous System Malformations
Disorders of the skull
Lückenschädel (“skull with gaps”)
Small posterior fossa
Low-lying tentorium cerebelli with large incisura
Scalloping of the petrous bone
Shortening of the clivus
Enlargement of the foramen magnum
Disorders of the cerebral hemispheres
Polymicrogyria
Cortical heterotopias
Dysgenesis of the corpus callosum
Large massa intermedia
Disorders of the posterior fossa
Descent of the cerebellar vermis through the foramen magnum
Caudal displacement of the pons and medulla
Rostral displacement of superior cerebellum through the tentorium
“Kinking” of the brainstem
Loss of pontine flexure
Aqueductal stenosis or forking
“Beaking” of the tectum
24.2 Epidemiology of Myelomeningocele, Prenatal Diagnosis, and Counseling
The incidence of MMC is approximately 0.7 to 0.8 per 1,000 live births, with regional variations. The rate in the United States is between 0.3 and 1.43 per 1,000 live births44,45 and is lower among African Americans than Caucasians. The incidence of MMC has declined46 because of improved maternal nutrition, folate supplementation, elective terminations, and other unknown factors. In the United States, the rate dropped from 0.6 to 0.3 per 1,000 live births between 1984 and 1992,44 with little additional decline as of 2004.47 The recurrence risk is 1 to 2% among parents having a single previously affected child,4,46 and 10% for those having two previously affected offspring.48 A woman with MMC has a 3% risk of bearing an affected child.4
Although these are most often isolated malformations, autosomal-dominant, autosomal-recessive, and X-linked–recessive transmissions exist.49 On the other hand, one study demonstrated a low concordance rate between monozygotic twins.50 Human NTDs are likely polygenic.
24.2.1 Prenatal Diagnosis
Maternal serum alpha fetoprotein screening is performed at 16 to 18 weeks of gestation and is 75% sensitive for detecting open NTDs (anencephaly and/or MMC).51 Underestimated gestational age, multiple pregnancies, and other fetal disorders, such as omphaloceles, can produce false-positive results; closed, skin-covered spinal malformations, such as myelocystoceles, may also be missed. Fetal ultrasound confirms the diagnosis with a sensitivity approaching 100% in recent series.51 Direct visualization of the placode and bony abnormalities, or indirect cranial signs such as the “lemon” and “banana” signs (▶ Fig. 24.2a,b) may be seen; the lemon sign has a sensitivity of 80% and the banana sign a sensitivity of 93%, with a false-positive rate of 0.88%.51 Amnionic alpha fetoprotein, acetylcholinesterase, and chromosomal analysis may be obtained via amniocentesis. Amnionic alpha fetoprotein levels are elevated in 97% of patients with NTDs,52 but acetylcholinesterase measurements are 14% more sensitive than alpha fetoprotein.51 False-positive elevations of both may occur with fetal blood contamination during sampling, impending fetal death or autolysis, and fetal abdominal wall defects51; false-negatives are seen with skin covered malformations.
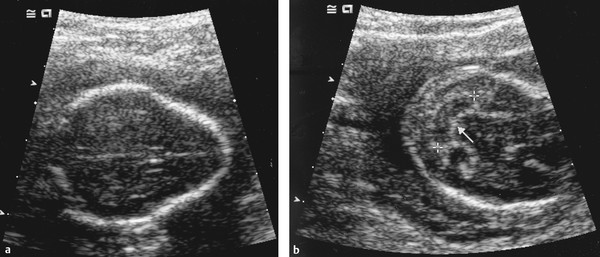
Fig. 24.2 Prenatal cranial ultrasound signs associated with myelomeningocele. (a) Lemon sign (convex inward frontal bones). (b) Banana sign (Chiari malformation).
24.2.2 Prenatal Counseling
Prenatal counseling should deliver accurate information upon which parents can make informed decisions. Almost all children survive with proper treatment53,54; deaths in infancy are most commonly caused by the Chiari malformation and in later life by unrecognized shunt malfunction. The overall long-term mortality is between 24 and 60%, with a mortality of between 15 and 34% in the first 5 years and of 9 to 26% thereafter.55,56
A normal IQ is attained by 70 to 75%56,57; some studies suggest a lower IQ for those with hydrocephalus.58–66 Sixty percent of those with a normal IQ have learning disabilities.60,67,68 Despite many challenges, 82% of adults are independent in activities of daily living and 30% attend or have finished college, but only 32% are gainfully employed.69 Achievement in one study was attained by 89% of those who never had a shunt placed, 69% of those with shunts but no revisions, 50% of those with shunt revisions before 2 years of age, and 18% of those having shunt revisions beyond 2 years of age.65
Ambulation correlates with sensorimotor level.70 Excluding 13% with severe delays, 89% of preadolescent children are community ambulators, including most with lumbosacral lesions and 63% of those with upper lumbar lesions.67 Ambulation falls to below 50% in adolescence as wheelchair use becomes more efficient.58 Few patients with lesions at L3 or a higher level are functional long-term ambulators.71
Urinary social continence (in social situations) is achieved in about 80 to 85% of school-age children.53,56,72–74 CIC is reported in 85%.56 Complete bowel continence is achieved in 50% of patients, and almost 90% report continence 75% or more of the time.56 About two-thirds of boys report at least some genital sensation, and 70 to 75% achieve reflex erections.
Approximately 70% of children require a ventricular shunt.53,58,67 Shunt malfunction occurs in 30 to 40% within 1 year, 60% within 5 years, and 85% within 10 years,58,75 and shunt infection occurs in 11 to 26%.76–78 One-third of children will undergo a tethered cord release, 15 to 33% a Chiari decompression, and 1 to 5% an operation for syringomyelia. Orthopedic and urologic procedures are also frequently required.
Cervicothoracic MMCs deserve special mention because the prognosis for sensorimotor function in these children is not predicted by their anatomical level. Most have no identifiable sensorimotor abnormalities at birth, although subtle impairments may become apparent over time; most develop hydrocephalus. Fetal magnetic resonance (MR) imaging is recommended before parents are counseled, especially if termination is being considered.79
The best mode of delivery is not clear; studies have produced conflicting results.80–88 Although two nonrandomized studies showed better outcomes following cesarian delivery,80,82 five other nonrandomized studies showed no consistent benefit to cesarian over vaginal delivery except perhaps in cases with a breech presentation or significant hydrocephalus.81,84–87 Finally, parents should be counseled about the increased risk for MMC with subsequent pregnancies and the need for periconceptional folate (4,000 µg of folate daily, 10 times the amount recommended for the general population).35
24.3 Neurosurgical Management of Children with Myelomeningocele
Given the complexity of MMC, some have thought it more merciful to withhold treatment and allow these infants to die. In 1971, Lorber developed criteria to select infants for nontreatment; these included severe hydrocephalus at birth, total paraplegia, spinal kyphosis, and an additional birth defect.89 In 2001, the Groningen Protocol was developed in the Netherlands to select infants for active euthanasia who were thought to be in “unbearable suffering” and to have a “hopeless prognosis”; 22 infants were euthanized by lethal injection as of 2010.
However, none of these criteria, either in isolation or in combination, are entirely accurate predictors of outcome,90 and in the United States, most children are treated. In a landmark study by McLone, the outcome of nonselective treatment compared favorably with that in the selected series of Lorber.91 Parents of children with MMC in the United States rarely regret a decision to treat their child, with only 4 of 300 regretting their decision to treat and 9 regretting their initial decision not to treat.90 Withholding treatment may rarely be considered for children with other, potentially fatal congenital malformations or chromosomal abnormalities, but this decision should be individualized and made only after a frank and realistic discussion with parents.
Neurosurgical care for the child with MMC can be divided into three phases: (1) initial stabilization of the infant and closure of the MMC; (2) assessment and treatment of associated hydrocephalus when present; and (3) long-term management of associated shunt malfunctions, the Chiari malformation, tethered spinal cord, and syringomyelia.
24.3.1 Stabilizing the Infant and Closing the Myelomeningocele
The MMC contains potentially functional tissue, so it should be protected from external mechanical trauma and desiccation and kept as clean as possible. Sterile saline-soaked gauze dressings and plastic wrap keep the malformation moist; iodine-containing compounds should be avoided as they may injure exposed tissues. Prophylactic antibiotics (usually ampicillin and gentamicin) are usually administered for the first 3 days.
The initial newborn evaluation should focus on several aspects. A general physical examination may disclose evidence of a more widespread genetic or developmental syndrome. Sensorimotor function is assessed by observing spontaneous leg movements and the response to pinprick stimulation (▶ Table 24.1). Muscle imbalance yields characteristic leg postures, such as hip flexion for upper lumbar level lesions, knee extension for midlumbar lesions, and ankle dorsiflexion with low lumbar level lesions. Clinical signs of hydrocephalus include accelerated head growth, full fontanel, split cranial sutures, and limitation of upgaze/“setting sun” eyes. Brainstem signs include bradycardia, apnea, dysfunctional swallow, high-pitched or weak cry, vocal cord palsies, and hypotonia. Severe brainstem dysfunction on the first day of life is a particularly ominous sign, especially if it persists after ventricular shunting.
| Level | Last intact motor level | Sensory level |
| T10 | Rectus abdominis | Umbilicus |
| L1 | Hip flexion | Anterolateral thigh |
| L2 | Hip adduction | Anteromedial thigh |
| L3 | Knee extension | Knee, anterior shin |
| L4 | Foot dorsiflexion | Dorsum of foot |
| L5 | Extensor hallucis longus | First–second interspace |
| S1 | Plantar flexion | Plantar aspect of foot |
The MMC may be repaired within the first 3 days without an increased complication risk.92 The placode is sharply dissected from the surrounding skin at the perimeter of the malformation (▶ Fig. 24.3a), avoiding injury to subjacent nerve roots. Skin tissue is meticulously dissected from the placode to avoid a subsequent inclusion dermoid.93,94 Small feeding arteries and draining veins are spared if possible. The lateral edges of the placode are approximated with the use of fine suture to reconstitute the neural tube (▶ Fig. 24.3c), which may make subsequent untethering easier to perform. Some surgeons excise the placode in patients with thoracic or thoracolumbar lesions and no leg function to facilitate dural closure and reduce the risk for subsequent retethering, but urologic worsening may occur in 8%.95,96
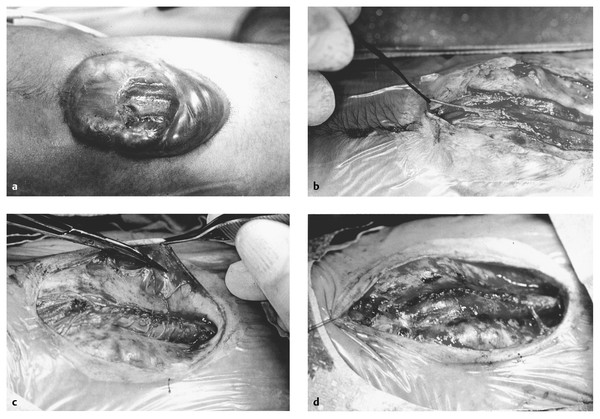
Fig. 24.3 Closure of myelomeningocele. (a) Myelomeningocele sac on dorsum of newborn infant. Placode is in the center and attached circumferentially to the adjacent dysplastic skin. (b) The placode has been dissected circumferentially from the skin and rolled up to re-create a neural tube. In this case, there is an intact filum terminale caudal to the placode, which will be sectioned. (c) The dura is dissected from under the skin circumferentially and brought together to cover the newly “closed” placode. (d) The dura is closed with running suture. The skin is undermined just above the lumbodorsal fascia and brought together in the midline with as little tension as possible. Another layer may be added by elevating the muscles and fascia on either side of the splayed posterior elements and bringing them together over the closed dura before skin closure.
Any other tethering malformation, such as a lipoma, split-cord malformation, or filum terminale, should be untethered if present (▶ Fig. 24.3b). One important variant, a hemimyelomeningocele, is suggested preoperatively by a significantly asymmetric motor examination and a sac that is slightly off midline. A hemimyelomeningocele is a form of split-cord malformation in which the spinal cord splits into two “hemicords,” one of which is exposed on the surface while the other is hidden under, and separated from, the exposed hemicord within its own separate dural sac.97
Once the placode is closed and all related tethering malformations treated, the surrounding dura is then circumferentially elevated from the underlying fascia (▶ Fig. 24.3c), and the dural sac is reconstituted with enough space to avoid strangulating the placode (▶ Fig. 24.3d). Dural grafts are rarely necessary, do not appear to decrease the risk for subsequent tethering, and may increase the risk for CSF leak.
The lumbodorsal fascia may be closed as an additional layer, but the benefits are unclear as cerebrospinal fluid (CSF) leaks occur in only 1 to 3% overall.98 One study found no additional benefit to myofascial closure,54 whereas a second study reported that CSF leakage and shunting were less frequent.99 Periosteal “turnover flaps” with the use of lumbodorsal fascia and acellular dermal matrix are also reported.100,101
Finally, the redundant skin is trimmed and closed. Vertical, horizontal, and Z-plasty closure techniques102 have been described, although a simple vertical closure is preferred because it simplifies later spine procedures should they become necessary. The skin is undermined circumferentially above the lumbodorsal fascia with sharp dissection; small perforating vessels arising from the fascia should be avoided if possible to prevent flap ischemia. As the skin edges are drawn together, some temporarily blanching often occurs but is usually transient; topical nitroglycerin paste may improve skin blood flow. Excess dysplastic skin is trimmed, and the normal skin is closed in a layered fashion.
Large skin defects may require rotational or myocutaneous flaps that are beyond the scope of this chapter and are reviewed elsewhere.103 Wound complication rates are lower following flap closure in some studies,104,105 but not others.106 In a novel technique (Ken Winston, personal communication, 1997), Silastic sheeting (used for creating silos for gastroschisis) is sewn circumferentially to the underside of the elevated skin flaps well back from the skin edges. The Silastic sheet is then plicated by drawing up the center of the sheet and suturing it every other day on the ward to progressively pull the flaps together; once the skin is overlapping, the Silastic sheet is removed and the wound closed primarily (▶ Fig. 24.4a,b). A similar progressive purse string cerclage technique has also been reported.107
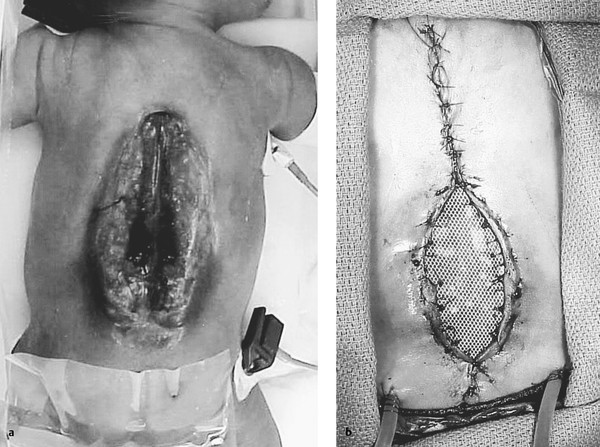
Fig. 24.4 Closure of large myelomeningocele by the progressive approximation of Silastic sheeting. (a) Huge thoracolumbar myelomeningocele. (b) Much of the myelomeningocele has been closed primarily, but the portion that remains open is attached along its undersurface to Silastic sheeting. This sheeting is plicated in the midline every few days in the neonatal intensive care unit, with the skin edges drawn progressively closer until they overlap. The child is then returned to the operating room, the Silastic sheet is removed, and the skin is closed primarily.
Severe kyphosis complicates about 12% of cases of MMC.108 Although most patients are paraplegic, a few have some preserved sensorimotor function. Kyphosis can lead to difficulty with the initial MMC closure, subsequent skin breakdown or ulceration, sitting imbalance, pulmonary restriction, and loss of neurologic function. Kyphectomy at the time of initial closure has been recommended because kyphosis commonly progresses, and later correction is risky. Kyphectomy in infancy significantly decreases recurrence, particularly if the lordotic curvature is restored.109 The placode is excised and the superior dura imbricated, or the placode and thecal sac are displaced laterally to expose the underlying vertebra(e) at the apex of the kyphos. Laterally displaced laminae and pedicles are removed with rongeurs. The vertebral bodies and adjacent disks at the apex of the kyphotic deformity are removed piecemeal with rongeurs and/or a high-speed drill, with extreme care taken to avoid injury to the subjacent vascular and retroperitoneal structures. Hemostasis is meticulous, but a transfusion should be anticipated. The kyphos is reduced by extending the femora under the drapes and bringing the adjacent end plates together; the newly approximated vertebrae are sutured together with several heavy (No. 1 or No. 2) nonabsorbable sutures supplemented with the excised bone chips.109 A body cast or custom orthosis is worn for 12 weeks.
24.3.2 Initial Treatment of Hydrocephalus
Hydrocephalus occurs in 80 to 90% of children with MMC,58,110 and 70 to 90% undergo shunting.56,111 For infants with overt signs of hydrocephalus at birth, MMC closure and contemporaneous shunting may be performed with no apparent increased risk and perhaps decreased wound leakage.112–114 External ventricular drainage or a ventricular reservoir may be used to temporize, particularly for those infants whose MMC was repaired more than 72 hours after delivery, in whom contemporaneous shunting is associated with a higher infection rate.115
For those with modest ventricular enlargement and no clinical evidence of hydrocephalus, neither absolute criteria nor consensus is available about when to place a shunt. One study suggested that infants with a cortical mantle thickness of 2.8 cm or larger may not need shunting.116 Various measurements, such as the Evans ratio or the average of the maximum frontal and occipital horn widths divided by the distance between the cortical surfaces,117 have been proposed but are rarely used in decision making. There is significant variability among pediatric neurosurgeons regarding the threshold for shunt placement in asymptomatic children with ventriculomegaly,118 likely related to uncertainty about the effects of moderate ventriculomegaly on brain development compared with the risks of shunt revisions and infections. Many pediatric neurosurgeons prefer moderate ventriculomegaly to ongoing and repeated shunt complications.118 On the other hand, cognitive improvements may occur even when a shunt is placed years later.119
24.3.3 Long-term Neurosurgical Management of Children with Myelomeningocele
Children with MMC are best managed in the setting of a multidisciplinary clinic specializing in the treatment of spina bifida, where pediatric surgical specialists, therapists, and a nurse coordinator are available120; the clinic can be an integral part of the medical home for these children.121 Pediatric neurosurgical care focuses on four major issues: the shunt, the Chiari malformation, syringomyelia, and spinal cord tethering. Management follows seven basic principles:
Each child has a unique set of static neurologic, orthopedic, and urologic deficits.
Each child has a set of radiographic abnormalities, including hydrocephalus in 70 to 80%, Chiari malformation in more than 95%, syringomyelia in 40 to 80%, and tethering in virtually 100%.
The mere presence of a radiographic abnormality, or even a radiographic change, does not necessarily direct treatment. Rather, treatment should generally be reserved for clinical deterioration.
Any neurologic, urologic, or orthopedic deterioration should prompt a search for a treatable cause.
Any clinical deterioration can potentially be caused by shunt malfunction, and all evaluations should therefore begin by excluding shunt malfunction.
If available, ventricular size at the time of suspected shunt malfunction must be compared with a baseline. Moreover, a shunt malfunction may occur with little or no change in ventricular size. An open mind is required.
Papilledema may occasionally develop with few or no symptoms; a funduscopic examination should be performed once the child is able to cooperate.
Shunt Management in Children with Hydrocephalus
The signs and symptoms of shunt malfunction in the child with an MMC are legion. The usual symptoms and signs—headache, nausea and vomiting, poor feeding, listlessness or lethargy, setting sun eyes, and extraocular abduction palsies—are not universal. Symptoms may be subtle or intermittent in nature, and of course not every headache is shunt-related. Experienced parents may be able to tell whether or not their child’s symptoms suggest shunt malfunction122; be wary of dismissing an experienced parent’s concerns! Other patterns of shunt malfunction include the following:
Cognitive changes such as a decline in school performance or worsening behavior
New onset of seizures or change in the pattern of seizures without another cause
Decreased arm or leg strength, loss of previously acquired motor skills, or increased tone
Unexplained change in ambulation
Worsening urinary and/or bowel function
Neck pain, lower cranial nerve or brainstem dysfunction.
Pain at the MMC closure site
Progressive scoliosis or orthopedic deformities
Any deterioration should first prompt a thorough investigation of shunt function before any other neurosurgical treatment is undertaken, especially if there are any associated headaches or cognitive changes.
Most children have increased ventricular size on computed tomography (CT) compared with their baseline. A shunt series may reveal disconnected or broken tubing. Notably, shunt pumping is of no demonstrated value.123,124 Other adjunctive measures may include a shunt tap, radionuclide study, or continuous intracranial pressure (ICP) monitoring. A shunt tap may be helpful if it demonstrates (1) sluggish or absent proximal flow (suggesting a proximal occlusion), (2) brisk flow and high pressure with manometry (suggesting a distal occlusion), or (3) proximate relief of headaches after fluid removal. Radionuclide shunt studies involve injecting tracer through the shunt into the ventricles while occluding distal outflow. Over time, the tracer should flow distally; failure to do so suggests malfunction. ICP monitoring is helpful if it demonstrates an acute rise in ICP concurrent with headaches. Unfortunately, shunt taps, radionuclide studies, and ICP monitoring are least reliable with a partially occluded shunt, and no test can absolutely exclude a shunt malfunction. Ultimately, the diagnosis rests on the clinical condition of the child, and a shunt exploration may be needed if symptoms continue. This cannot be overemphasized—nowhere else in pediatric neurosurgery is clinical judgment so important, and misjudgment so treacherous.
Endoscopic third ventriculostomy (ETV) has emerged as an alternative to shunt revision with overall success rates of higher than 70% among eligible patients with MMC and (1) evidence of noncommunicating hydrocephalus by CT ventriculography and/or MR imaging, (2) minimal or no discernible subarachnoid space, and (3) a third ventricle at least 4 mm wide.125 The success rates are lower in children younger than 6 months than in older children.125 ETV is technically more challenging and difficult in this population because of abnormal third ventricular anatomy, distorted foramina of Monro, and interthalamic adhesions. A combination of ETV and choroid plexus cautery (ETV/CPC) was more successful (76%) among infants with MMC in sub-Saharan Africa than was ETV alone (35%), with comparable intellectual outcomes and fewer later failures.126,127 Whether these results can be reproduced remains to be seen.
Can a child with MMC and hydrocephalus managed with a shunt ever be considered shunt-independent? There are certainly anecdotes of shunt disconnections or breaks seen incidentally on radiographs in asymptomatic patients. However, abrupt or even fatal deterioration has been reported months or years later.128–130 A radionuclide study may confirm that CSF is not flowing through the intervening shunt tract,131 but the validity of this technique has not been evaluated in patients with MMC. The difficulty in determining shunt independence with certainty, and the possibility of sudden decompensation, should be carefully discussed if observation is chosen.
Management of the Chiari Malformation
The Chiari 2 malformation is a pancerebral abnormality (see box “Chiari-Associated Central Nervous System Malformations”) that is present in most children with MMC. Herniation of the cerebellar vermis, brainstem, and fourth ventricle through the foramen magnum may cause lower brainstem signs and symptoms, although most children are relatively asymptomatic and fewer than one-third require surgery.58,91,132–137 Presenting symptoms are most frequent during the first year, with lower cranial neuropathies, swallowing dysfunction, and disordered breathing predominating.132 Almost all children with MMC have modest swallowing issues, such as a hyperactive gag reflex, intolerance for particular food textures, gagging (usually on solids), and more frequent vomiting, all of which usually improve over time and do not require treatment. More concerning are progressive swallowing abnormalities, such as choking on liquids, significant weight loss, nasal regurgitation, and aspiration pneumonia. Other concerning signs may include those of disordered breathing: central or obstructive apnea, cyanotic spells (sometimes even when the child is on a ventilator133), inspiratory stridor, and a hoarse, weak, or high-pitched cry. Stridor and apnea at delivery are more ominous and have a higher mortality rate and poorer outcome than stridor and apnea that develop later.138 Vocal cord palsies, particularly when bilateral, are often irreversible and require tracheostomy.139 Other signs and symptoms (particularly in the older child) may include weakness and/or spasticity of the upper extremities, pain (headache and/or neck pain), cerebellar problems, oculomotor dysfunction, and scoliosis.42,132,139,140
The Chiari 2 malformation is well seen on sagittal T1 or T2 MR images (▶ Fig. 24.5). A speech and swallow evaluation may disclose phonation difficulties, hoarseness, or abnormal swallow. A formal swallowing study132 may disclose disordered swallowing mechanics, incomplete clearing or pooling of tracer in the vallecula, occult tracheal aspiration, or reflux. Radionuclide swallow studies may also reveal occult aspiration. Direct laryngoscopy may disclose unilateral or bilateral vocal cord weakness, and pulmonary studies may demonstrate episodic central or obstructive apnea.
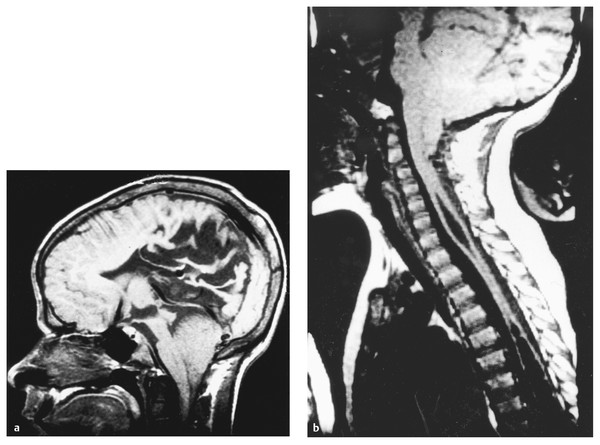
Fig. 24.5 Chiari 2 malformation and syringomyelia. (a) Midsagittal T1-weighted magnetic resonance (MR) image shows disordered sulcation of the posterior cortex (stenogyria), dysgenesis of the corpus callosum, beaked tectum, and enlarged massa intermedia of the thalamus. The cerebellar vermis and brainstem lie below the foramen magnum. (b) Midsagittal T1-weighted MR image showing descent of the cerebellar vermis and brainstem below the foramen magnum and two syringes in the midcervical and thoracic regions.









