Myoclonic Epilepsies Associated with Progressive Degenerative Disorders: Progressive Myoclonic Epilepsies
Myoclonic Epilepsies Associated with Progressive Degenerative Disorders: Progressive Myoclonic Epilepsies
The progressive myoclonic epilepsies (PMEs) have received considerable attention in the neurologic literature (Genton et al, 2000; Roger et al., 1992; Berkovic et al., 1986; Diebold, 1973) even though, numerically, they represent only a small fraction of the myoclonic epilepsies. Aicardi and Chevrie (1971) found only nine patients with PMEs in a study of 90 children and adolescents with myoclonic seizures. Aicardi (1980b) extended this series to 145 patients, and found only 11 who had a progressive encephalopathy. Because these figures come from a specialized referral center, they certainly overestimate the proportion of severe and unusual cases, so the true figures would be even lower. The interest aroused by PME, however, is justified by the genetic origin of these disorders, the severity of their prognosis, and the insight that inborn errors of metabolism can provide into the normal functioning of the neuronal chemical machinery.
Following the early descriptions by Unverricht in 1881 and Lundborg from 1903 to 1913, the concept of PMEs as a single individualized and recognizable syndrome became firmly established. The PME syndrome is characterized by the following four main elements: (a) myoclonic jerks, which are segmental, fragmentary, and usually erratic in topography; (b) epileptic seizures, mainly generalized tonic-clonic or clonic-tonic-clonic seizures and massive myoclonic seizures; (c) progressive mental deterioration; and (d) variable neurologic signs and symptoms, mainly cerebellar, extrapyramidal, and action myoclonus (Fig. 7.1). Diebold (1973) separated the myoclonic epileptic syndromes with dementia into two groups. In the first group, the myoclonic epileptic syndrome is a constant and central characteristic of the causal disorders (e.g., Lafora bodies disease). In the second group, the myoclonic epileptic syndrome is a manifestation of the causal disorder only occasionally (e.g., in Huntington chorea).
More recently, the value of the concept of PME has been increasingly questioned. The PME syndrome clearly is comprised of a heterogeneous collection of unrelated disorders that are only superficially similar. The mechanism of the myoclonus itself varies with the causes, and neurophysiologic studies permit the separation of the several mechanisms responsible for the jerks. The remaining usefulness of the concept of PMEs is to offer a broad framework that permits temporary accommodation of some as yet unclassified conditions featuring epileptic seizures, myoclonus, and neurologic degeneration.
Most disorders responsible for PME with or without dementia have relatively characteristic clinical, electroencephalographic (EEG), and other features that permit individual recognition (Aicardi, 1982b), and enzymatic assays, linkage and/or deoxyribonucleic acid (DNA) studies, and/or peripheral tissue biopsies are available to confirm the diagnosis.
This chapter therefore describes the major conditions that are responsible for PMEs in infancy, childhood, and adolescence. Cases in young adults that pose the same problems as those in adolescence are included. However, most disorders that produce myoclonus in infancy and early childhood are quite different from the traditional picture of the PME syndrome in older children and adolescents, and they raise completely different diagnostic problems. These are dealt with only briefly, and the interested reader is referred to their descriptions in specialized articles (Genton et al., 2002; Livet et al., 2002; Delgado-Escueta et al., 2001; Berkovic et al., 1986, 1993). The late infantile type of ceroid lipofuscinosis is one such case, because differentiating it from the early myoclonic epilepsies and Lennox-Gastaut syndrome (LGS) is difficult, thus exposing this problem, especially since it has no real resemblance to the traditional PME syndrome (Harden and Pampiglione, 1982). Similarly, the late type of ceroid lipofuscinosis may pose difficult diagnostic dilemmas with some disorders of vision and/or hysteria, not with the PMEs, because the myoclonus is usually a late manifestation.
The main conditions that give rise to PMEs are listed in Table 7.1. In this table, the disorders have been divided into three groups. The first group includes those disorders that produce a relatively typical PME syndrome, as described earlier in this chapter. The second group includes the diseases that give rise to myoclonic epilepsies that clearly differ from those in the first group in their associated signs. The third group comprises a variety of disorders that are associated with myoclonic jerks and other epileptic manifestations but that differ considerably from the classic PME syndrome in age at onset and clinical and EEG manifestations. A complete description of the conditions listed in Table 7.1 is, however, beyond the scope of this book.
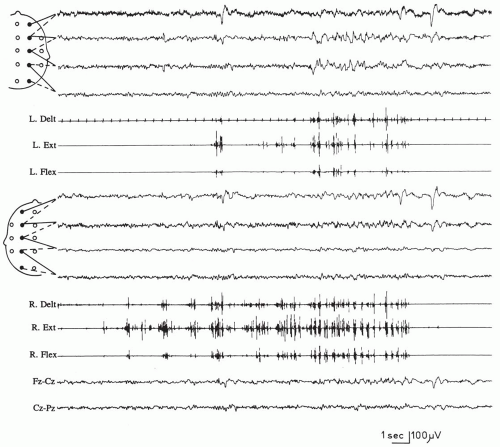
FIG. 7.1. A 27-year-old woman with sialidosis (cherry-red spot myoclonus) and movement-activated seizures. When the patient raises her right arm to seize a hammer, a typical “cascade” of myoclonic potentials that are initially arrhythmic and that involve all of the sampled muscles on the right (right deltoid and wrist extensors and flexors) occurs; these then become more rhythmic, spreading to the contralateral muscles. This pattern of spread of myoclonic activity is typical of action myoclonus.
Most of the conditions listed are genetic diseases that are mostly inherited as autosomal recessive traits. Their geographic and ethnic distribution varies. For example, Unverricht-Lundborg disease is common in Finland (Norio and Koskiniemi, 1979) and relatively common in northern European populations, whereas Lafora disease is most prevalent in the Mediterranean Basin and several Middle and Far East countries. However, extensive overlap does exist (Delgado-Escueta et al., 2001).
In some PMEs (e.g., Gaucher disease, several types of ceroid lipofuscinosis), the metabolic error responsible for the disease is known, permitting both a confirmation of the diagnosis and reliable prenatal diagnosis. Linkage studies have defined 12 loci of the PME syndromes. More recently, DNA studies have led to the discovery of the mutated genes of several PMEs, including Lafora disease, Unverricht-Lundborg disease, dentatorubral-pallidoluysian atrophy (DRPLA), myoclonic encephalopathy with ragged-red fibers (MERRF), and five of the ceroid lipofuscinoses (Table 7.2).
TABLE 7.1.Diseases that may cause progressive myoclonic epilepsy
Group I:typical or relatively typical progressive myoclonic epilepsy syndrome
Lafora disease
Unverricht-Lundborg disease
Juvenile neuronopathic Gaucher disease
Galactosialidosis
Dentatorubral-pallidoluysian atrophy
Juvenile neuroaxonal dystrophy
Group II:atypical progressive myoclonic epilepsy syndrome
Aug 1, 2016 | Posted by drzezo in NEUROLOGY | Comments Off on Myoclonic Epilepsies Associated with Progressive Degenerative Disorders: Progressive Myoclonic Epilepsies