Fig. 1
Rabies virus (RABV) tricky travel. Propagation of RABV particles (green dots) from the site of entry (muscle) up to the brainstem and the exit site (salivary glands) is represented by the green arrows. RABV is a strictly neurotropic virus causing no viremia. In the periphery, innate (type 1 IFN) and adaptive (T, B neutralizing antibodies) immune responses are triggered by the detection of virus particles in the muscle. After entry into the nerve, RABV infection has to face two host’s traps: the premature death of the infected neuron and the launch of an innate immune response by neurons and surrounding glial cells. This local innate immune response results in the production in the nervous system (NS) of antiviral, chemoattractive and inflammatory molecules which facilitates the infiltration of immune cells (monocytes, T and B cells) from the periphery. These cells have the potency to eliminate the infected neurons. Finally, RABV encounters the immune responses once again at the end of its cycle when it is directed into the salivary glands by centrifugal passage. The dark blue frame represents the immunological privileged sites of the NS. The light blue frame represents the extraneural peripheral tissues and organs (including the immune system in the periphery)
However, despite these robust lines of defence, once a virulent RABV has entered the NS its progression is not interrupted. This might be the result of the intrinsic capacity of this virus to evade the innate immune response launched by the infected neurons (Rieder and Conzelmann 2009) to preserve the integrity of the infected neurons (Prehaud et al. 2010) but also to eliminate the protective T cells migrating into the NS (Lafon et al. 2008). In addition, RABV may also benefit from the intrinsic capacity of NS to control the immunological homeostasis of the body resulting in a down-regulation of the immune responsiveness in the periphery (Camelo et al. 2001) (Fig. 2).
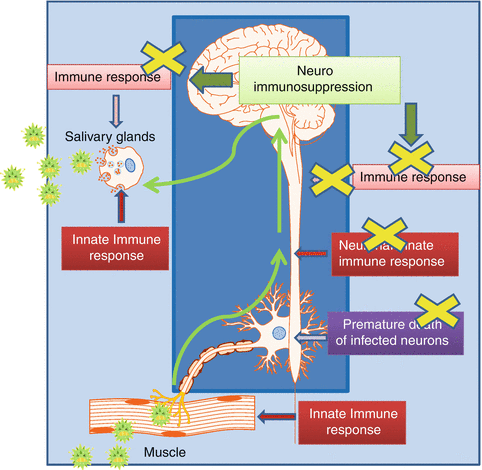
Fig. 2
Rabies virus (RABV) spoils host’s plans to dampen the virus progression. RABV has evolved strategies to escape the host defenses and complete its cycle in the infected host. These host defenses can exert their protective functions at the site of entry and possibly also at the final stage of infection, in the salivary glands. However, they might be down-regulated by central neuro-immuno-suppression
The knowledge we have gained of the interactions of RABV with the immune responses has been obtained mainly from models of experimental rabies in mice using laboratory-adapted RABV strains after intramuscular or intraplantar (footpad) route of administration to mimic natural transmission by a bite. Comparison of the features of pathogenic strains causing fatal encephalitis in mice and attenuated strains responsible of abortive non-fatal disease has allowed elucidating of which parameters control RABV pathogenicity.
2 RABV Preserves the Integrity of the Infected Neurons
The capacity to kill or injure infected cells is a widespread property of viruses. Nevertheless, a few viruses such as RABV are able to grow without inflicting major damage to the host cells. Analysis of autopsied rabies-infected patients indicates that despite heavy viral load, neuronal apoptosis is a rare event in natural rabies (Jackson et al. 2008). Experimentally, in a model of RABV infection of non-human primates 4 days after infection, infected motor neurons show no signs of degeneration with normal size, morphology and Nissl staining (Ugolini 2010). In vitro, rat spinal motoneurons never encounter death (Guigoni and Coulon 2002). Nevertheless, later in the infection, when the brain is heavily infected, degenerative changes are seen in dendrites and axons, and peripheral nerve dysfunction occurs (Juntrakul et al. 2005; Li et al. 2005; Scott et al. 2008; Kojima et al. 2009; Rossiter et al. 2009; Jackson et al. 2010). Thus, it can be stated that successful RABV propagation in the NS requires that neuronal cell bodies are not damaged by premature apoptosis and that the integrity of axons and dendrites is preserved, at least during the period of time required for the virus to reach the brain.
Only attenuated RABV strains used for vaccines (such as the Evelyn Rotniki Abelseth (ERA) strain) or RABV related viruses of bat origin (such as Lagos bat virus) trigger apoptosis (Thoulouze et al. 1997; Kassis et al. 2004). Induction of apoptosis may contribute to the high immunogenicity of live rabies vaccines (Megret et al. 2005). M protein (Lagos bat virus) and the cytoplasmic domain of the G protein (in the case of the attenuated RABV ERA) have been found to control the commitment of infected cells toward death (Larrous et al. 2010; Prehaud et al. 2010; Babault et al. 2011).
Virulent RABV strains not only preserve the integrity of the infected neurons but also actively promote neuron survival (Prehaud et al. 2010). The capacity of RABV to promote neuronal survival or death depends upon the cellular partners recruited by the PDZ-binding site (PDZ-BS) of its envelope G protein. Neuronal survival requires selective association of the PDZ-BS with the PDZ domain of two closely related serine-threonine kinases (MAST1 and MAST2). However, a single amino acid change in the PDZ-BS that triggers neuronal death, allows G protein to recruit additional PDZ partners, notably a tyrosine-phosphatase, PTPN4. Silencing this phosphatase abrogates RABV-mediated apoptosis. PDZs are globular domains that play a central role in cell signaling by favoring spatial contacts between enzymes and their substrates. Disruption of the complexes formed by PDZ and the PDZ-BS encoded by their ligands may have profound effect on cellular signaling pathways—MAST2 functioning as an inhibitor of neuronal survival and PTPN4 as an inhibitor of cell death. It is likely that RABV G proteins antagonize PTPN4 and MAST2 functions by simply disrupting the interaction of PTPN4-PDZ and MAST2-PDZ with their respective cellular ligands (Prehaud et al. 2010; Babault et al. 2011; Terrien et al. 2012).
3 RABV Innate Immune Response
3.1 Innate Immune Response in the Periphery
The innate immune response is the first line of defence against infectious agents. It involves the release of type 1 interferons (IFN-α/β), inflammatory cytokines and chemokines, the activation of complement and the attraction of macrophages, neutrophils and NK cells into infected tissues. This innate immune response is triggered in the first hours following the entry of pathogens and is not pathogen specific. It contrasts with the adaptive immune response that is tailored to a specific pathogen and requires several days to develop. The innate immune system can sense the presence of micro-organisms through “pattern recognition receptors” (PRRs) that recognize danger signals and “pathogen associated molecular patterns” (PAMPs) expressed by microbes. Toll like receptors (TLRs) or retinoic acid inducible gene (RIG) like receptors (RLRs) are important PRRs for the recognition of viral double-stranded RNAs and single-stranded RNAs (Liu et al. 2008; Hornung et al. 2006). Resulting signal transduction cascades involve TRIF, Myd88 or IPS-1 as adaptors of TLR3, or TLRs other than TLR3 and of RLRs, respectively.
Once RABV is inoculated in the skin or in muscle by bites or scratches, the presence of the virus is rapidly detected by host defence mechanisms. The IFN response triggered at the site of entry has an antiviral effect. This was demonstrated by comparing the viral load in the thigh muscle of two groups of mice, parental mice and mice lacking the type I IFN receptor (IFNAR) after injection in the hindlimb with the encephalitic RABV strain, CVS. The viral load measured by the accumulation of viral RNA was increased in mice lacking IFNAR compared to parental strain of mice (Chopy et al. 2011a). This observation suggests that some viral particles might be readily eliminated at this early step of infection.
3.2 Innate Immune Response in the NS
Like most tissues in the organism, the NS expresses different types of PPRs capable of sensing dangerous and pathogen signals (Boivin et al. 2002; Nguyen et al. 2002; Bottcher et al. 2003; Koedel et al. 2004; McKimmie et al. 2005). Central neurons express TLR1-4 as well as TLR 7 and 8 (Prehaud et al. 2005; Ma et al. 2006; Kim et al. 2007; Ma et al. 2007; Tang et al. 2007; Barajon et al. 2009). These express the RLRs (RIG-I and Mda-5) (Lafon et al. 2006; Menager et al. 2009; Peltier et al. 2010; Chopy et al. 2011b), but not the RLR LGP2, which seems to be actively degraded in neurons (Chopy et al. 2011b). Peripheral nerve plexuses and nerves (dorsal root ganglion sensory neurons and fibers of sciatic nerves) express TLRs (TLR3, 4 and 7) with prominent expression of TLR3 (Cameron et al. 2007; Barajon et al. 2009; Goethals et al. 2010). Neurons take an active part in the innate immune response in the brain being both responders to IFN and IFN producers, secreting type I IFN (predominantly IFN-β in the brain, no IFN-α and no Type III IFN λ) (Prehaud et al. 2005; Delhaye et al. 2006; Sommereyns et al. 2008).
RABV is known to trigger a RIG-I-mediated innate immune response in neurons (Hornung et al. 2006) by detecting the 5′tri-phosphate base pairing of the viral genome (Pichlmair et al. 2006). After infection, human neurons can mount a classical primary IFN response (activation of IRF3 and NF-kappa B), as well as a secondary IFN response, (activation of STATs and IRF7), leading to the production of cytokines (IL-6, TNF-alpha) and chemokines (CXCL10 and CCL5) (Prehaud et al. 2005; Chopy et al. 2011a, b).
3.2.1 RABV Dampens the IFN Response in the Infected Neurons
Viruses have evolved sophisticated strategies to escape the innate immune response (Randall and Goodbourn 2008; Versteeg and Garcia-Sastre 2010). This is the case for RABV (for review see Rieder and Conzelmann 2009). The N and the P protein of RABV are multifunctional proteins involved in RNA synthesis and in counteracting the host innate immune response. The N protein limits RIG-I-signaling (Masatani et al. 2010, 2011), whereas the P protein inhibits IRF3 and IRF7 phosphorylation (Brzozka et al. 2005; Rieder et al. 2011), suppresses STAT1 nuclear translocation (Brzozka et al. 2006; Vidy et al. 2007) and sequesters an antiviral protein, the promyelocytic leukemia (PML) protein, in the cytoplasm (Blondel et al. 2010). As a result, down-regulation of the IFN response can be observed in vitro. For example, in RABV-infected human post-mitotic neurons (NT2-N), transcription of IFN-β gene is seen as early as 6h post infection, and IFN-β protein is produced during the first 24 h post infection, whereas, transcription and production decline thereafter (Prehaud et al. 2005).
It has been shown that virulence, at least for a Japanese vaccine strain (Nishigahara RABV strain), depends upon the capacity of this strain to evade the innate immune response and this process is controlled by the ability of the P and N protein to evade the innate immune response (Shimizu et al. 2006; Masatani et al. 2010). Thus, evasion of the IFN response in infected neurons may be critical for RABV progression in the NS through the neuronal network allowing the virus to reach the brainstem and the salivary glands.
3.2.2 RABV Limits the Inflammatory Response in the NS
Inflammation is a key component of host responses to cell damage or microbial entry leading to the production of inflammatory mediators including complement, adhesion molecules, cyclo-oxygenase enzymes and their products, as well as cytokines or chemokines. Release of these toxic factors has dramatic consequences when the site of inflammation is the NS, where severe dysfunction can lead to significant NS pathology with neuronal death (Brown and Neher 2010). In the brain, both neurons and glial cells can mount antiviral, inflammatory and chemokine responses. Astrocytes can respond to the presence of innate immune stimulus in the brain by producing proinflammatory cytokines and chemokines (Park et al. 2006). Nevertheless, an important role is taken by microglia in the induction of neuro-inflammation, a feature, which may reflect the density or the sub cellular localization of the innate immune receptors (Bsibsi et al. 2006).
Transcriptome and proteomic analysis of the inflammatory response triggered in the NS of mice by various virulent strains of RABV showed that RABV infection stimulates the expression of chemokines (CCL5, CCL2, CCL9 and CXCL9) and inflammatory cytokines (IL-6, IL-12) (Camelo et al. 2000; Baloul et al. 2004; Wang et al. 2005; Chopy et al. 2011b; Sugiura et al. 2011). When compared with other encephalitic virus infections such as caused by Borna virus, RABV triggers only limited inflammation (Shankar et al. 1992; Fu et al. 1993). Moreover, the inflammatory reaction in the RABV-infected NS is transient with the expression of a majority of markers being rapidly down-regulated in the spinal cord and with a slight delay in the brain (Chopy et al. 2011b).
Comparison of the inflammatory reaction triggered by RABV strains of various degree of pathogenicity indicates that the more pathogenic strains trigger weaker inflammatory responses (Baloul and Lafon 2003; Wang et al. 2005; Laothamatas et al. 2008; Hicks et al. 2009).
It is likely that this low inflammatory reaction in the infected NS contributes to keeping intact the BBB, a condition that correlates with RABV pathogenicity, with non-pathogenic RABV strains triggering a transient opening of the BBB, but not pathogenic strains (Phares et al. 2006; Roy et al. 2007).
Limitation of neuroinflammation by virulent RABV occurs by several mechanisms. RABV infection avoids neuronal apoptosis (Lafon 2011) and rarely infects glial cells; two intrinsic features of the infection contributing to limit neuroinflammation. In addition, in the course of RABV infection the expression of anti-inflammatory molecules is up-regulated in the NS. This is the case for the anti-inflammatory soluble proteins TNFR1 and 2, which can interfere with the binding of TNF to its receptors (Chopy et al. 2011b). This is also the case for the suppressors of cytokine signaling (SOCS), a family of proteins that negatively control cytokine signal transduction, with SOCS-1 being up-regulated in the brain of RABV infected dogs in non infected cells in close vicinity of infected neurons (Nuovo et al. 2005). More importantly, RABV up-regulates the expression of HLA-G, a non-classical MHC molecule and B7-H1, the ligand of PD-1, (programmed death protein-1), in neurons, and also in the infected NS, in the case of B7-H1 (Lafon et al. 2005, 2008). Besides, their immune-tolerant properties, which are exploited by RABV, (see below Sect. 4), HLAG and B7-H1 molecules are now also considered as providing negative feedback that limits tissue inflammation (Carosella et al. 2001; Phares et al. 2010). This, in particular, is the case for B7-H1, which dampens the expression of pro-inflammatory molecules (such as iNos and TNF-α) during viral encephalitis (Phares et al. 2010), whereas, HLA-G influences the cytokine balance towards a Th2 pattern by promoting the secretion of IL-4, IL-3 and IL-10 and down-regulating the production of IFN-γ and TNF-α (Carosella et al. 2001).
Limitation of inflammation by RABV infection might result from (1) reducing the entry in the NS of mononuclear leukocytes, monocytes and macrophages, (2) maintaining the impermeability of the BBB, and (3) minimizing the release of neurotoxic molecules that can compromise NS function and host survival.
These conditions should preserve not only the integrity of the infected neuronal network, but also the life of the host, allowing the virus to reach the brainstem and the salivary glands before the premature death of the infected host.
4 RABV Adaptive Immune Response
Mounting an adaptive immune response against a microbe, even a neurotropic virus that rapidly enters the NS after its inoculation in muscle, always occurs in the periphery and never in the NS, which is devoid of lymphoid organs (Galea et al. 2007). The triggering of the adaptive immune response that takes place in the lymphoid organs such as the lymph nodes or spleen relies on the activation of plasmacytoid dendritic cells (DCs), pDCs and of type 1 IFN that they produce in a TLR7 and 9 dependent manner after encountering the microbe (Diebold et al. 2003; Steinman 1991). In natural rabies infection, the nature of cells producing type I IFN at the site of the injection is not known with certainty. Candidates are muscle cells, fibroblasts, keratinocytes, dendritic cells (DCs) and macrophages (Charlton and Casey 1979, 1981). There is experimental evidence that RABV can infect bone marrow derived conventional DCs (cDCs) and macrophages in vitro. Despite non-productive infection, RABV triggers the production of IFNs, cytokines and chemokines in these cells (Nakamichi et al. 2004; Faul et al. 2010). Infection of sentinel cells may be dispensable since macrophages activation was observed in vitro when inactivated RABV was added to the culture (Nakamichi et al. 2004). In cell culture, maturation of cDCs in the presence of RABV is controlled by type 1 IFN whose production might rely on the recognition of intra cytoplasmic RABV RNAs through RIG-I and mda-5 receptors and not TLR7 (Faul et al. 2010), a characteristic of cDCs (Eisenacher et al. 2007).
4.1 RABV Specific Immune Response in the Periphery
After the injection of the CVS strain of rabies in the hind limb of mice, the size of the draining popliteal lymph nodes and those of spleen increase. Draining lymph nodes are populated with activated T cells expressing the marker of activation CD69. Activation can also be observed among peripheral blood lymphocytes (Vuaillat et al. 2008).
A strong B cell response is mounted in the spleen. When mice were injected with a less pathogenic virus (the PV strain), similar activation of T cells was observed in lymph nodes and blood suggesting that adaptive immune response is independent of the virulence of the RABV strain. Indeed, when mice were injected with an encephalitic RABV bat strain (silver-haired bat rabies virus, SHBRV) or with a less pathogenic virus (CVS-F3, mutant of CVS encoding a mutation in the G protein), the resulting adaptive immune responses (neutralizing antibodies, CD4+, CD8+ T cells response) were not different (Roy and Hooper 2007). This suggests that adaptive immune response triggered by RABV strains in the periphery, an event that occurs late after the virus has entered the NS, is unrelated to RABV pathogenicity. This may explain why patients die of rabies despite having mounted an immune response in the periphery attested by the presence of neutralizing antibodies in the blood (Hunter et al. 2010). Nevertheless, in an experimental model of rabies in skunks, a cyclophosphamide-induced immunosuppression was found to reduce the infection of the salivary glands (Charlton et al. 1984), suggesting that the adaptive immune response may control to some extent the final stage of RABV infection. This control may also be exerted on viral particles at the site of entry if entry into the NS has been delayed.
4.2 RABV Provokes the Killing of Migratory T Cells
Infiltrating T cells control most of the infections of the NS. This is, for example, observed during the course of West Nile virus brain infection, where CD8+ T cells attracted by the chemokines produced by inflammatory cells in the infected NS are a critical factor for controlling the infection (Klein et al. 2005; Zhang et al. 2008). In rabies, sterilisation of the infection by T cells is inefficient, and their activities are specifically inactivated by the virus (Lafon 2008). Immunohistochemical studies performed on autopsy materials obtained from deceased rabies-infected patients revealed that the cells undergoing death were leukocytes and not neurons (Hemachudha et al. 2005; Tobiume et al. 2009). This observation was reproduced in mice infected with the encephalitic RABV strain CVS. Immunocytochemistry of brain and spinal cord sections revealed that despite a heavy load of viral antigens, infected neurons do not undergo apoptosis. In contrast, the migrating T cells (CD3+) were apoptotic (Baloul and Lafon 2003; Baloul et al. 2004; Lafon 2005a; Kojima et al. 2009; Rossiter et al. 2009). Moreover, pathogenicity of the CVS strain was similar in immunocompetent mice Balb/c mice and in Nu/Nu Balb/c mice, indicating that T cells do not control the outcome of encephalitic rabies (Lafon 2005a). In striking contrast, deprivation of T cells transformed an abortive infection into a encephalitic rabies similar to that caused by the encephalitic strain CVS infection, showing that T cells is a critical factor in the restriction of the NS infection caused by an abortive RABV strain. Indeed, when apoptosis was analyzed in the spinal cord of immunocompetent mice infected with the abortive RABV strain PV, killing of T cells was not observed; instead, infected neurons died (Galelli et al. 2000). Altogether, these observations indicate that T cells have a protective potential to control RABV infection in the NS, nevertheless their capacity to control RABV infection is impeded with the encephalitic RABV strain. The mechanisms by which the encephalitic RABV strain evades the host T cell response was further studied as described below.
Tumors evade immune surveillance by multiple mechanisms, including the inhibition of tumour-specific T cell immunity. In order to escape attack from protective T cells, tumor cells up-regulate expression of certain surface molecules such as B7-H1, Fas-L, and HLA-G, which triggers death signalling in activated T cells expressing the corresponding ligands PD-1 for B7-H1, Fas for FasL and CD8—among others—for HLA-G (Gratas et al. 1998; Dong et al. 2002; Rouas-Freiss et al. 2003). Studies evaluating whether RABV-infected neurons up-regulate immunosubversive molecules to kill activated T cells following an evasive strategy similar to that selected by tumors cells have been undertaken both in vivo and in vitro. In vitro, RABV infection was found to up-regulate the expression of HLA-G at the surface of human neurons (Lafon et al. 2005; Megret et al. 2007). In vivo, comparison of experimental rabies in mice caused by CVS, which kills T cells, or by PV, which does not kill T cells, led to the finding that the CVS-infected NS, but not the PV-infected NS, up-regulates the expression of FasL. In mice lacking a functional FasL, there was less T cell apoptosis in the NS than in control mice. Remarkably, RABV morbidity and mortality were reduced in these mice. Destruction of T cells through the Fas/FasL pathway can be enhanced by indoleamine 2, 3 dioxygenase (IDO), the expression of which in the infected neurons and the brain is up-regulated by RABV (Prehaud et al. 2005; Zhao et al. 2011). The enzyme IDO converts extracellular tryptophan into kynurenine, thereby reducing its concentration in the microenvironment that in turn markedly enhances the sensitivity of any nearby T cell for Fas-ligand induced apoptosis (Kwidzinski et al. 2003).
In addition, RABV-infected brain up-regulates the expression of another immunosubversive molecule, B7-H1 (Lafon et al. 2008). Whereas non-infected NS was almost devoid of B7-H1 expression, RABV infection triggers neural B7-H1 expression that increases as the infection progresses. Infected neurons and also other non-infected neuroectodermal cells, including astrocyte-like cells, were found positive for B7-H1. RABV infection of B7-H1 deficient mice resulted in a drastic reduction in clinical signs and mortality. Reduction of RABV virulence in B7H1−/− mice was concomitant of a reduction of CD8+ T cell apoptosis among the migratory T cells.
Thus, despite the triggering of a classical adaptive immune response in the periphery and the infiltration of the lymphocytes into the infected NS, the protection, which could have been conferred in the NS by this immune response, is drastically impeded by RABV infection.
5 RABV Infection Triggers a CNS Mediated Immune-Unresponsiveness
The dampening of immune protection already triggered by RABV is completed by a central immunosupression caused by the neuronal reflex control of immunity triggered by the NS facing an excess of inflammation in its attempt to restore general homeostasis.
RABV infection by a pathogenic strain induces an immune-unresponsiveness (Wiktor et al. 1977a, b; Torres-Anjel et al. 1988; Perry et al. 1990; Hirai et al. 1992; Tshikuka et al. 1992; Kasempimolporn et al. 1997, 2001; Camelo et al. 2001) characterized by the impairment of T cells functions with an alteration of cytokine pattern, an inhibition of T cells proliferation and the destruction of immune cells without modification of immune cells proportion (CD4/CD8 ratio constant) in the lymphoid organs (Perry et al. 1990). This leads to the atrophy of the spleen and the thymus of RABV infected mammals. TNF-α receptor has been found to play a role in RABV immune-unresponsiveness, since immune cells lacking the TNF-α p55 receptor were less immunosuppressed compared to the wild-type (Camelo et al. 2000). Importantly, infection of the brain is required since immune-unresponsivenesss does not occur after the infection of the NS with an abortive RABV strain, which infects the spinal cord only (Camelo et al. 2001). This suggests that the property of the NS that centrally controls the immune response in the periphery might be triggered (Tracey 2009). NS modulates the immune functions through two main immune-neuroendocrine pathways: the hypothalamo-pituitary-adrenal (HPA) axis and the autonomic NS (ANS) composed of sympathetic and parasympathetic nerves fibres (Johnston and Webster 2009). The homeostatic reflex is activated after the brain senses the presence of an excess of inflammatory cytokines such as TNF-alpha, IL-1β or IL-6 in the periphery, by neuronal (mainly through local afferent fibres of the vagus nerve) and by humoral pathway (Johnston and Webster 2009) processed by the NS in hypothalamic and brainstem centres.
Related posts:
 in Leprosy: Insights from Model Systems and Patients
Health Problems Associated with the Use and Abuse of Khat (Catha edulis)
Neurology of a Permanent and Non-progressive Motor Neuron Disorder Associated with Food (Cassava) Toxicity
Preservation, Snake Venoms and Stroke in the Tropics
Infections: Neurological Involvement and Neurobiology
Neurological Involvement
in Leprosy: Insights from Model Systems and Patients
Health Problems Associated with the Use and Abuse of Khat (Catha edulis)
Neurology of a Permanent and Non-progressive Motor Neuron Disorder Associated with Food (Cassava) Toxicity
Preservation, Snake Venoms and Stroke in the Tropics
Infections: Neurological Involvement and Neurobiology
Neurological Involvement
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


