CHAPTER 26
Neuroendocrinology
I. Hypothalamus
A. Hormones that affect pituitary function
1. Corticotropin-releasing hormone: mainly from the paraventricular nucleus; stimulates adrenocorticotropic hormone (ACTH); stimulated by stress, exercise; inhibited by glucocorticoids through negative feedback
2. Thyrotropin-releasing hormone: a tripeptide secreted mainly from the paraventricular nucleus; stimulates thyroid-stimulating hormone (TSH) and prolactin; decreased by stress, starvation, and thyroid hormones through negative feedback
3. Gonadotropin-releasing hormone (GnRH): secreted mainly from the arcuate nucleus; pulsatile release stimulates follicle-stimulating hormone (FSH) (slower pulse frequency) and luteinizing hormone (LH) (more rapid pulse frequency); continuous exposure to GnRH actually decreases luteinizing hormone and FSH through down-regulation; negatively affected by stress, low body weight, weight loss, excessive exercise (which cause hypothalamic amenorrhea); clinical application: treatment of precocious puberty of hypothalamic origin makes use of long-acting GnRH agonists (through down-regulation).
4. Growth hormone (GH)–releasing hormone: a peptide secreted from the arcuate nucleus; stimulates GH; clinical application: recombinant human GH replacement therapy is given for GH deficiency.
5. Somatostatin or somatotropin release-inhibiting factor: a peptide secreted mainly from the periventricular nuclei; also from the gastrointestinal tract; inhibits release of GH; clinical application: somatostatin analogues (e.g., octreotide and lanreotide) and GH receptor antagonists (pegvisomant) are used as adjuncts in treatment of acromegaly (GH excess).
6. Dopamine: from the arcuate nucleus; inhibits release of prolactin; prolactin inhibitory factor; suppression, not stimulation of prolactin release, is the major hypothalamic effect on prolactin; clinical applications: destruction of the hypothalamic-pituitary connection (such as transection of the pituitary stalk) produces a decrease in the release of pituitary hormones, except for prolactin, which is increased because dopamine (prolactin inhibitory factor) is the major regulator of this pituitary hormone; dopamine agonists such as bromocriptine and cabergoline are used in the treatment of prolactin-producing tumors.
B. Appetite
1. The hypothalamus has mediators or receptors for mediators of food intake.
a. For increased appetite: ghrelin, neuropeptide Y
b. For satiety or reduced food intake: leptin, cholecystokinin, serotonin (lorcaserin, a selective serotonin agonist, is an appetite suppressant)
2. Areas of the hypothalamus that control eating
a. Lateral nuclei = feeding center; lesions in this area produce adipsia, aphagia.
b. Ventromedial nuclei = satiety center; lesions in this area produce hyperphagia.
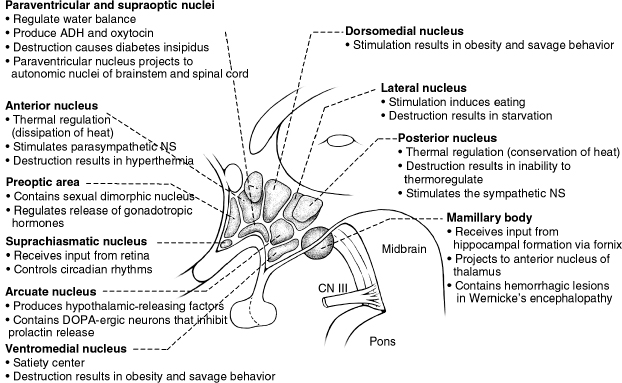
Figure 26.1 The hypothalamus with its various nuclei and corresponding functions. ADH, antidiuretic hormone; CN, cranial nerve; NS, nervous system.
C. Emotion/behavior: stimulation of the septal region results in feelings of pleasure and sexual gratification; lesions in the caudal hypothalamus produce attacks of rage; impaired GnRH release causes decreased libido; the opioid peptides enkephalin and dynorphin are involved with sexual behavior.
D. Temperature
1. Pre-optic anterior hypothalamus: lesions of this area produce hyperthermia.
2. Posterior hypothalamus: lesions of this area produce hypothermia and poikilothermia.
II. Pituitary
A. Anterior pituitary (adenohypophysis) hormones
PITUITARY HORMONE | EXCESS (ADENOMAS) | DEFICIENCY |
ACTH | Cushing’s disease | Adrenal insufficiency (glucocorticoid axis) |
TSH | Hyperthyroidism | Hypothyroidism |
FSH and LH | Usually silent | Infertility, hypogonadism |
GH (somatotropin) | Gigantism in children; acromegaly in adults | GH deficiency |
Prolactin | Amenorrhea, galactorrhea | Inability to lactate |
Abbreviation: LH, luteinizing hormone.
1. Pituitary tumors: 30% to 40% are prolactinomas; 20% are somatotropinomas; 10% to 15% are corticotropinomas; 1% are thyrotropinomas; 25% are clinically nonfunctioning (includes gonadotropinomas)
a. Hyperprolactinemia: produces amenorrhea, galactorrhea, low testosterone levels in males
i. Causes: prolactinomas: more than 70% are microadenomas (<10 mm); the rest are macroadenomas (>10 mm);
ii. Diagnosis: prolactin levels, pituitary MRI
iii. Treatment of choice: dopamine agonists—bromocriptine, cabergoline
Physiologic | Pregnancy |
Sleep | |
Nursing | |
Stress | |
Nipple stimulation | |
Drugs | Dopamine receptor blockers |
Antipsychotics, especially first generation | |
Opioid analgesics | |
Estrogens | |
α-Methyldopa | |
CNS lesions | Prolactinomas |
Lesions of the hypothalamus or pituitary stalk, granulomatous disease | |
Others | Liver cirrhosis |
Chronic renal failure | |
Primary hypothyroidism (via TRH stimulation) |
Abbreviations: CNS, central nervous system; TRH, thyrotropin-releasing hormone.
b. Acromegaly: causes frontal bossing, coarse facial features, increased shoe and ring size, carpal tunnel syndrome, hyperhidrosis;
i. Diagnosis: elevated GH and insulin-like growth factor-1; lack of GH suppression after oral glucose tolerance test; pituitary MRI
ii. Treatment of choice: transsphenoidal surgery; adjuncts: radiation, dopamine agonists such as bromocriptine (because dopamine attenuates GH secretion in one-third of patients), GH receptor antagonist (pegvisomant), somatostatin analogues (octreotide, lanreotide)
c. Cushing’s disease: Cushing’s syndrome due to a pituitary adenoma (other causes of Cushing’s syndrome are exogenous glucocorticoid intake, adrenal tumors, and ectopic ACTH production); presents with moon facies, buffalo hump, purple striae, diabetes, centripetal obesity; diagnosis: screening by 1-mg overnight dexamethasone suppression test, 48-hour low-dose dexamethasone suppression test, midnight salivary cortisol, or by urinary-free cortisol
i. To differentiate from adrenal causes: ACTH, corticotropin-releasing hormone test; to differentiate from ectopic causes: pituitary MRI, inferior petrosal sinus sampling
ii. Treatment: transsphenoidal pituitary surgery is the treatment of choice; other treatments: mifepristone (glucocorticoid receptor antagonist for glucose intolerance) and pasireotide (analog of somatostatin receptor subtype 5 which is overexpressed in corticotroph adenoma cells).
d. Thyrotropinomas: rare; manifests with hyperthyroidism (symptoms include palpitations, nervousness, weight loss, increased appetite, increased sweatiness), diffuse goiter; diagnosis: TSH levels are normal or high, thyroxine (T4) and triiodothyronine (T3) levels are high (as opposed to hyperthyroidism from a thyroid origin such as Graves’ disease, in which TSH is low while T4 and T3 are high); elevated α subunit levels; pituitary MRI; macroadenoma in 90% of cases.
i. Treatment: surgery is the treatment of choice; adjuncts are radiation, somatostatin analogs such as octreotide, or treatment targeted toward the thyroid gland itself, such as antithyroid drugs, radioactive iodine ablation, or thyroidectomy.
e. Gonadotropinomas: rare, usually clinically silent
2. Hypopituitarism: may be inherited or acquired (e.g., from compression, inflammation, invasion, radiation of the hypothalamus or pituitary); for acquired disorders: prolactin deficiency is rare and occurs only when the entire anterior pituitary is destroyed (e.g., pituitary apoplexy) (remember that tonic inhibition by dopamine is the predominant control of prolactin); of the remaining cells, the corticotrophs and thyrotrophs are usually the last to lose function.
a. Adrenal insufficiency: affects the glucocorticoid, not the mineralocorticoid, axis (for adrenal insufficiency originating from the adrenals, both glucocorticoid and mineralocorticoid axes are affected); presents acutely with hypotension, shock; chronic adrenal insufficiency presents with nausea, fatigue
i. Diagnosis: ACTH stimulation test (however, will not differentiate between primary adrenal failure and secondary pituitary failure)—draw baseline cortisol levels, administer synthetic ACTH (e.g., Cortrosyn®), 250 µg intramuscularly or intravenously, then draw cortisol levels again at 30 and 60 minutes; normal if peak cortisol is greater than or equal to 18 to 20 µg/dL; may give false normal results in acute cases because the adrenal glands may still produce cortisol; in the acute setting, do not need to wait for lab values to come back before instituting glucocorticoid treatment if clinically warranted; in acute stressful situations, hydrocortisone has conventionally been given at a total daily dose of 300 mg intravenously, but lower doses are also effective; maintenance treatment is usually with hydrocortisone, 20 mg in the morning and 10 mg at night, or prednisone, 5 mg in the morning and 2.5 mg at night, or less if tolerated.
b. Hypothyroidism: not apparent acutely because the half-life of serum T4 is approximately 7 days
i. Diagnosis: normal or low TSH, low T4 and T3 (as opposed to primary hypothyroidism, in which TSH is high)
ii. Treatment: glucocorticoids should be replaced before thyroid hormone replacement; replacement is with levothyroxine preparations such as Synthroid®
c. Hypogonadotropic hypogonadism: delayed puberty, amenorrhea in females: can be seen in female athletes; low testosterone levels in males (causes sexual dysfunction, decreased libido)
i. Treatment: delayed puberty: testosterone for boys, estrogen for girls; luteinizing hormone-releasing hormone or FSH and human chorionic gonadotropin to induce ovulation/fertility; treatment with testosterone replacement in adults: intramuscular or topical preparations; monitoring of prostate-specific antigen levels (link with prostatic cancer though causality not yet proven) and complete blood count (can cause polycythemia)
ii. Inherited disorders
(A) Kallmann’s syndrome: hypogonadotropic hypogonadism, anosmia
(B) Laurence-Moon-Biedl: autosomal recessive, hypogonadotropic hypogonadism, mental retardation, obesity, retinitis pigmentosa, syndactyly
(C) Prader-Willi: hypogonadotropic hypogonadism, hyperphagia, obesity, mental retardation; caused by loss of function in region of chromosome 15; most cases occur when segment of paternal chromosome 15 is deleted in each cell.






