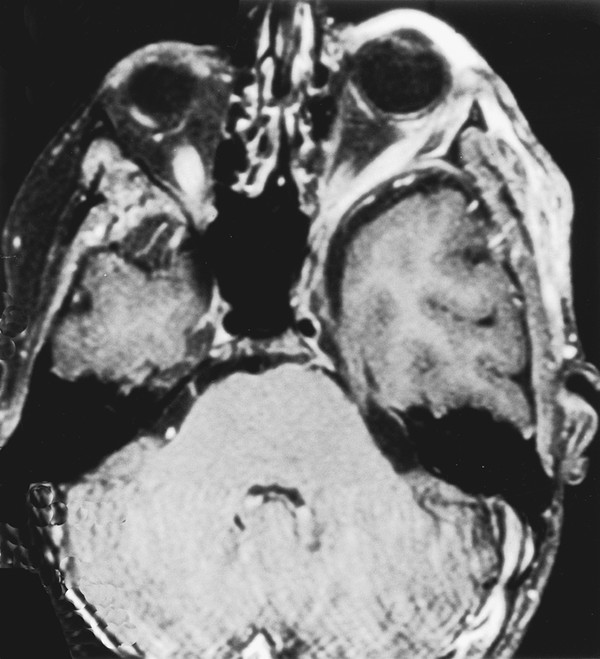
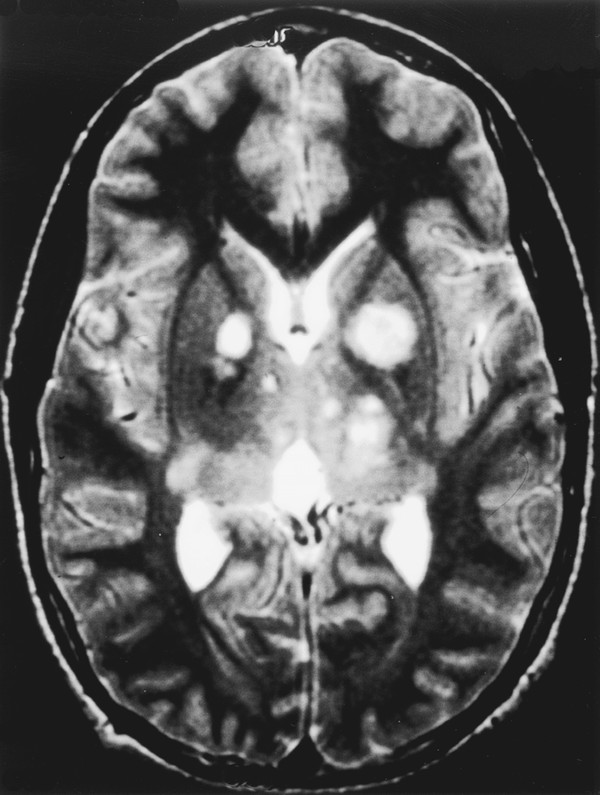
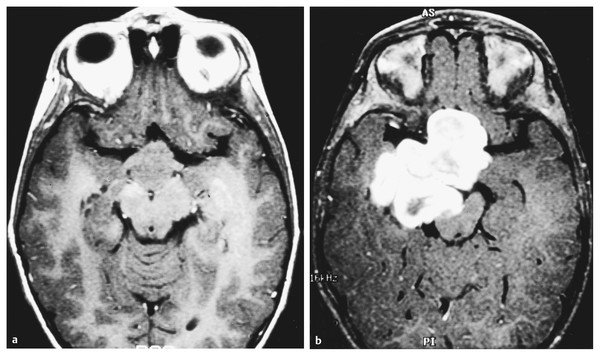
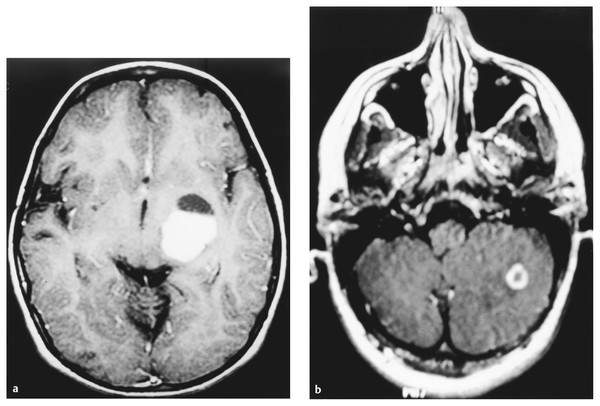
Neurofibromatosis is a descriptive term that was coined by Frederick von Recklinghausen in 1882 to characterize the cutaneous tumors of two patients that were thought to be composed of a combination of neural and mixed cellular elements. This diagnostic category was later expanded to incorporate an array of dermatologic, ocular, and nervous system manifestations; thus, it was included with the broader group of disorders referred to as phakomatoses. In the ensuing decades, investigators realized that patients with NF could be categorized into distinct clinical groups based on the pattern of neural and extraneural manifestations.1 Improvements in imaging techniques and advances in molecular genetics have facilitated these efforts and allowed clinicians to confidently classify patients as having neurofibromatosis 1 (NF-1, previously known as peripheral neurofibromatosis) or neurofibromatosis 2 (NF-2, previously known as central neurofibromatosis), and they have also identified phenotypically similar variants that are distinct entities.2,3 The importance of recognizing these disorders stems from the fact that the natural history of a neoplasm, such as a peripheral nerve tumor or an optic glioma, may be significantly different depending on whether or not the lesion arises in a person with NF. In addition, the indications for therapeutic intervention, the hierarchy of treatment options, and the long-term management goals may differ substantially for patients with NF-related versus sporadic tumors. Finally, recognition of the diagnosis is an essential step in providing appropriate multidisciplinary evaluation and counseling to affected patients and their families. This chapter focuses on the diagnostic and therapeutic issues that arise in children with NF-1 and NF-2; other phakomatoses are discussed in the following chapter. NF-1 is one of the most common genetic disorders, affecting 1 in 3,000 to 1 in 4,000 people.4,5 The mode of inheritance is autosomal-dominant, and approximately 50% of cases arise sporadically as new mutations. The syndrome results from mutations or deletions of a gene on chromosome 17q11.2 that encodes a large protein called neurofibromin. A portion of this protein is a guanosine triphosphatase (GTPase) activator6,7 that plays a role in signal transduction by favoring conversion of the active GTP-bound form of Ras and related G-proteins to the inactive guanosine diphosphate (GDP)–bound form.8 The NF-1 gene functions as a classic tumor suppressor gene in that loss of both alleles is needed for tumorigenesis. Because patients with NF-1 are born with only one normal copy of the gene, a single mutation or deletion that inactivates the second allele is theoretically sufficient to favor tumor formation,9 although additional molecular events may also contribute.10 Affected patients exhibit a combination of café au lait macules, Lisch nodules (iris hamartomas), axillary and inguinal freckling, skeletal lesions such as sphenoid wing dysplasia and thinning of long bone cortices, and optic gliomas, as well as an increased incidence of other central nervous system and systemic tumors.10–15 Diagnostic criteria that reflect the diverse manifestations of NF-1 were proposed at a National Institutes of Health (NIH) Consensus Development Conference16 (see box “NIH Consensus Criteria for the Diagnosis of Neurofibromatosis 1”16). They were devised, in part, for high specificity (i.e., to have a low rate of false-positive diagnoses). Because many of the characteristic stigmata, such as Lisch nodules, are not usually apparent in infancy, ongoing follow-up is sometimes required to establish the diagnosis conclusively. In a large study of patients with sporadic NF-1, 54% of children met diagnostic criteria by 1 year of age, 97% by 8 years, and 100% by 20 years.17 The exclusion of non–NF-1 variants, such as Proteus syndrome (the original “elephant man” disease that was mistaken for NF-1) and Legius syndrome, has been facilitated by genetic testing when clinically warranted.2 NIH Consensus Criteria for the Diagnosis of Neurofibromatosis 1 The diagnostic criteria are met if a patient has two or more of the following: Six or more café au lait macules that have a maximum diameter of more than 5 mm in prepubertal patients and more than 15 mm in postpubertal patients Two or more neurofibromas of any type, or one or more plexiform neurofibromas Freckling in the axillary or inguinal region Optic glioma Two or more Lisch nodules (iris hamartomas) A characteristic osseous lesion, such as sphenoid wing dysplasia or thinning of the long bone cortices, with or without pseudarthrosis A first-degree relative (i.e., parent, sibling, or child) with NF-1 by the above criteria Source: National Institutes of Health Consensus Development Conference. Neurofibromatosis. Conference statement. Arch Neurol 1988;45(5):575–578.16 NF-2 is less common than NF-1, affecting 1 in 25,000 to 1 in 50,000 people.18,19 This disorder reflects mutations or deletions involving a gene at chromosome region 22q12 that encodes a protein referred to as merlin (moesin-, ezrin-, and radixin-like protein); merlin is involved in linking cytoskeletal elements with plasma membrane proteins.20 Affected patients have a combination of eighth nerve and other cranial nerve neurilemomas, meningiomas, glial neoplasms, neurofibromas, and juvenile posterior subcapsular cataracts (see box “Consensus Criteria for the Diagnosis of Neurofibromatosis 2”16). As in patients with NF-1, the diagnosis in patients without a positive family history may be difficult initially because many children will not manifest sufficient findings to satisfy diagnostic criteria conclusively, and they may present at a young age with isolated nervous system tumors, such as spinal cord ependymomas and unilateral vestibular schwannomas.21,22 In such “suspected” cases, ongoing surveillance is warranted. Consensus Criteria for the Diagnosis of Neurofibromatosis 2 The diagnostic criteria are met if a person has either of the following: Bilateral eighth nerve masses seen with appropriate imaging techniques, such as magnetic resonance imaging or computed tomography A first-degree relative with neurofibromatosis 2 and a unilateral eighth nerve mass or two of the following: Neurofibroma Meningioma Glioma Neurilemoma Juvenile posterior subcapsular cataract Source: National Institutes of Health Consensus Development Conference. Neurofibromatosis. Conference statement. Arch Neurol 1988;45(5):575–578.16 A distinct subgroup of patients with features of NF-1 and NF-2 exhibit signs restricted to certain segments of the body. In the most typical situation, patients will have café au lait macules and neurofibromas on one extremity or one-half of the body and may have Lisch nodules in the ipsilateral eye. This so-called segmental form of NF-1 accounts for 5% of patients with NF-1 and arises from mosaicism, in which mutations of the NF1 gene occur at some time after fertilization in the developing embryo.3,13,23 If gonadal progenitors are spared (i.e., there is just somatic mosaicism), then this form of the disorder is not genetically transmissible. If both gonadal and somatic cells are involved, then a potential exists for genetic transmission, which varies from nearly zero (if a small percentage of gonadal cells are involved) to 50% (if all gonadal cells are involved). A segmental form of NF-2 has also been suggested for patients who have multiple discrete neurilemomas involving peripheral nerves of an extremity without central features of NF-2.23 Even among patients with typical (nonsegmental) NF-1 or NF-2, there is significant variability in the severity of manifestations between members of different families. The basis for this symptomatic heterogeneity may in part reflect differences in the specific site of mutations in the NF1 or NF2 genes themselves24: patients with large deletions involving the NF1 locus and surrounding genes tend to have a more severe phenotype than those with point mutations,25 whereas those with small in-frame gene deletions have a milder variant.26 The results of increasingly available genetic tests, such as protein truncation assays, fluorescent in situ hybridization, and direct sequencing, correlated with the clinical features, should shed further light on this issue during the next few years and should also help to resolve the diagnostic uncertainty that often surrounds patients who meet only one criterion for NF-1 or NF-2. However, an important caveat in interpreting these genetic studies is that the exact features of NF-1 and NF-2 can vary widely within a single family (in which all affected individuals should have an identical NF mutation), reflecting the likely involvement of other disease-modifying genes or interacting environmental factors. Thus, contemporary genetic testing can predict the occurrence of NF-1 or NF-2, but not its severity in most cases.24 Box “Suggested Screening Studies for Children with Proven or Presumptive Neurofibromatosis 1” and box “Suggested Screening Studies for Children with Proven or Presumptive Neurofibromatosis 2” summarize the baseline evaluations that are recommended in children with proven or suspected NF-1 or NF-2. Similar suggestions for health supervision in children with NF-1 have been published by the Committee on Genetics of the American Academy of Pediatrics.27 Such recommendations represent guidelines rather than requirements, and as such, they are followed with flexibility and judgment. They incorporate a detailed screening of the major systems involved by each of the disorders, particularly the skin, eyes, nervous system, and spine, and provide a basis for a more detailed evaluation of other systems if concerning findings are detected on a screening evaluation. For example, children with NF-1 or NF-2 do not undergo routine imaging of the chest and abdomen, but such studies are employed for those who present with symptoms of dyspnea, abdominal discomfort, or distention, which may be referable to an enlarging thoracoabdominal tumor. Similarly, a comprehensive endocrine evaluation coupled with cranial magnetic resonance (MR) imaging is pursued in children with NF-1 who manifest precocious puberty, growth delay, or other evidence of endocrinopathy that may be related to hypothalamic involvement by tumor. Finally, because children with NF-1 often exhibit varying degrees of cognitive impairment that may interfere with their school performance and socialization,28,29 detailed neuropsychological testing is often pursued, but the need for this testing is determined on a case-by-case basis. Suggested Screening Studies for Children with Proven or Presumptive Neurofibromatosis 1 Annual clinical examination including neurologic assessment and dermatologic evaluation Annual ophthalmologic examination Magnetic resonance (MR) imaging examination of the head Children diagnosed before 5 years of age Children with new neurologic deficits, visual loss, or endocrinopathy MR examination of the spine and plain radiographs Children with scoliosis Children with back pain, radiculopathy, or long tract signs referable to the spine Neuropsychological and developmental testing Children with learning, speech, or socialization difficulties or impaired fine motor skills Genetic counseling should be offered to the family at diagnosis and as needed on an ongoing basis. Suggested Screening Studies for Children with Proven or Presumptive Neurofibromatosis 2a Neurologic examination Ophthalmologic examination Audiogram Magnetic resonance imaging of the head and spine Genetic counseling should be offered to the family at diagnosis and as needed on an ongoing basis. aThe frequency with which these tests are repeated depends on whether abnormalities are identified on the initial evaluation. At the least, all tests should be repeated approximately every 3 to 5 years. Annual examinations are recommended for children with known lesions. An important element in the evaluation of such children is the need for a comprehensive approach to patient care, often best provided in the setting of a multidisciplinary clinic. This approach facilitates efforts to diagnose and treat the child as an individual rather than as a collection of affected organ systems and ensures that management and counseling proceed in a coordinated fashion. A discussion of the major nonneurologic and neurologic manifestations of these disorders and their diagnosis and management is provided below. Because both NF-1 and NF-2 are multisystem disorders, patients often present with symptoms and signs not directly referable to a nervous system tumor. Appropriate recognition of the significance of these findings and knowledge of the indications for diagnostic and therapeutic intervention are essential for optimizing functional outcome. Café au lait macules and axillary freckling are often a source of concern but are of no serious clinical significance. These lesions result from abnormal collections of melanin pigment in affected melanocytes, which harbor loss or inactivation of both NF1 alleles.30 Café au lait macules, in particular, can occur in a variety of syndromes other than NF-1; thus, their detection in an infant or young child does not signal the need for extensive neurodiagnostic imaging in the absence of other clinical stigmata or a family history of NF-1, but it does warrant conscientious pediatric follow-up. Similarly, Lisch nodules simply represent melanocytic iris hamartomas; although these lesions increase in frequency during childhood and are present in more than 90% of affected patients by the completion of puberty,17,31 they do not interfere with vision. In contrast, skeletal manifestations can be of major long-term functional significance. Congenital bowing and/or dysplasia of the long bones, particularly the tibia, may lead to pathologic fractures that resist healing.14,32 Osseous dysplasia can involve the sphenoid bone as a congenital or acquired process,33 which leads to herniation of the temporal lobe contents into the orbit and, in some cases, produces pulsatile proptosis and seizures (▶ Fig. 48.1). Because few patients exhibit progressive impairment from this deformity, operative intervention should be limited to those children with worsening proptosis in the absence of another explanation, such as an orbital plexiform neurofibroma, or with intractable seizures from the involved temporal lobe. In such rare cases, reconstruction with split-thickness calvarial grafts or rib grafts may be beneficial. Fig. 48.1 This axial T1-weighted magnetic resonance image shows left sphenoid dysplasia with protrusion of the temporal dura into the posterolateral orbit and resultant proptosis. Spinal manifestations are also common, even in the absence of neoplastic involvement. Some degree of scoliosis is present in most patients with NF-1,14,32 but it often does not require specific therapy. However, in a small percentage of children with NF-1, the scoliosis is severe and rapidly progressive. Because many of these patients will be found to have an intra- or extra-axial neurofibroma, which may need to be addressed in conjunction with a spine-straightening procedure, MR imaging is an essential step in the preoperative evaluation. Regardless of the cause for the scoliosis, the rapid progression that occurs in some children mandates that vigilant follow-up and expeditious intervention be pursued to avoid severe deformity. Patients often require a combination of bracing, anterior and posterior fusion, and instrumentation to treat this problem. In such cases, the use of MR imaging–compatible hardware is advisable because these patients generally require long-term surveillance for tumor growth. Other, less serious spinal manifestations include vertebral scalloping and nonneoplastic widening of the neural foramina from dural ectasia.34 Most cases require no specific intervention. An additional phenomenon that is occasionally observed in patients with NF-1 is segmental hypertrophy.5 This may involve a portion of the head or one of the extremities. Although there is usually an underlying neoplastic component in the involved area, the deformity often exceeds that directly attributable to the tumor. It remains uncertain whether this reflects generalized mesenchymal dysplasia in the involved area or a combination of neurogenic and humoral factors initiated by the tumor. Children with NF-1 have also been noted to have an increased incidence of central precocious puberty and growth hormone deficiency; in some cases, this is independent of any radiologically apparent hypothalamic tumor involvement. In addition to the nervous system tumors discussed below, patients with NF-1 are also at risk for a variety of systemic malignancies, including leukemia, pheochromocytoma, rhabdomyosarcoma, adenocarcinoma of the ampulla of Vater, melanoma, and non-Hodgkin lymphoma, presumably reflecting either loss or inactivation of the second NF1 allele or other secondary genetic events.5,35,36 In patients with NF-2, the major serious nonneural manifestation is the development of posterior subcapsular cataracts, which are detected in 85% of affected individuals and often progress with age.12 Because these lesions can threaten vision, conscientious ophthalmologic follow-up is required, and surgical removal of the cataract may be indicated. In patients with unilateral visual loss secondary to one of these lesions, particular attention must be directed to monitoring and protecting vision in the contralateral eye, which includes preserving facial nerve function to maintain eye closure and corneal protection. Children with either NF-1 or NF-2 may present with neoplasms of the brain, spinal cord, and peripheral nerves, but the most common types of lesions differ significantly in these two syndromes. In addition, patients with NF-1 may exhibit a variety of nonneoplastic neurologic manifestations that must be distinguished from tumors to avoid unnecessary intervention. The most common processes are summarized below, along with management approaches. By far the most common abnormalities on MR imaging in patients with NF-1 are foci of increased signal on T2-weighted images without mass effect or contrast enhancement, so-called unidentified bright objects (UBOs; ▶ Fig. 48.2). These foci, which are detected in 60 to 80% of children with NF-1,37,38 may be solitary, multiple, or confluent and are seen most commonly in the basal ganglia, internal capsule, brainstem, and cerebellum. In view of the high frequency of these lesions in patients with NF-1, it has been suggested that their presence be added to the diagnostic criteria for this syndrome. On serial images in individual patients, foci of T2 signal abnormality often increase in frequency and number early in childhood, then regress later in childhood.37,38 This pattern suggests that these lesions represent age-related abnormalities in myelination. Some groups have noted that the presence and extent of these signal abnormalities correlate with the detection of learning disabilities, which are encountered in at least 25% of patients with NF-1, although others have failed to note a clear relationship.28,29 Similarly, it remains uncertain whether patients with UBOs are at increased risk for the development of central nervous system neoplasia compared with patients who have NF-1 without this finding, although at least one group has noted such a trend.39 Fig. 48.2 This axial T2-weighted magnetic resonance image demonstrates the characteristic areas of T2 signal abnormality that are commonly detected within the basal ganglia and occasionally within the cerebellum and other regions of the brain in patients with neurofibromatosis type 1. Because UBOs themselves typically follow a benign course, their detection in an otherwise asymptomatic child does not signal the need for serial MR imaging. However, this conservative follow-up approach should not be applied to childhood lesions that exhibit atypical MR imaging features, such as mass effect or enhancement, or that are associated with focal neurologic symptoms. Such a follow-up approach should also not be followed in older patients with new lesions. In all these instances, the natural history remains uncertain. The second most common imaging abnormality in NF-1 is optic pathway glioma, detected in at least 15% of patients (▶ Fig. 48.3). Several characteristic lesion types may be seen. The mildest abnormalities consist only of thickening of one or both optic nerves40; although most such lesions are low-grade gliomas, others may simply represent hyperplasia of the optic nerve sheath. Other patients exhibit a globular thickening of the optic nerves and chiasm (▶ Fig. 48.3a) that may occur in conjunction with T2 signal abnormalities streaking backward along the optic pathways and upward into the hypothalamus. Biopsy of such lesions has generally confirmed the presence of a low-grade glioma.41,42 Finally, a small percentage of patients present with a large mass lesion involving the optic chiasm and hypothalamus that may extend upward into the third ventricle, laterally into the temporal fossa, anteriorly beneath the frontal lobe, and posteriorly into the perimesencephalic region (▶ Fig. 48.3b). Fig. 48.3 Two magnetic resonance images that illustrate the diverse manifestations of optic–hypothalamic gliomas in patients with neurofibromatosis type 1. (a) Globular enlargement of the optic chiasm is depicted. In this patient, T2 signal abnormality was seen along the optic tracts bilaterally. (b) A massive chiasmatic–hypothalamic glioma shows bright enhancement with intravenous contrast. The optimal management for such lesions remains controversial. Before the era of MR imaging and high-resolution computed tomography (CT), optic pathway tumors were generally detected only after the onset of visual impairment, hypothalamic dysfunction, or symptoms of increased intracranial pressure (ICP), which occur in a small percentage of patients and clearly mandate therapeutic intervention. However, most lesions are now detected in asymptomatic individuals in whom the natural history and the indications for intervention are less clear. Several studies have reported the results of expectant management in patients with NF-1 who had optic pathway tumors that were asymptomatic or minimally symptomatic in association with mild visual loss or precocious puberty.41–43 Listernik et al43 noted that only 3 of 33 asymptomatic or minimally symptomatic patients with optic pathway tumors exhibited progressive tumor growth or deteriorating vision after diagnosis, with a median follow-up of 2.4 years. Other groups have also noted that optic gliomas in children with NF-1 have a distinctly more indolent course than in children without this disorder,44,45 although a sizeable subset do progress radiologically and in terms of visual loss.46,47 Thus, although the detection of an initially asymptomatic or minimally symptomatic optic pathway lesion on screening MR imaging does not signal the need for immediate intervention, ongoing imaging surveillance is prudent until the natural history can be established conclusively. However, the role of routine imaging as a screening test remains controversial because most optic pathway lesions in patients with NF-1 are asymptomatic and show a low frequency of significant enlargement, at least in the span of several years.48 Several groups have recommended annual MR imaging studies in young children with NF-1. Others have limited imaging to patients with new or progressive symptoms and signs, such as nystagmus, strabismus, visual loss, visual field deficits, precocious puberty, growth delay, diencephalic syndrome, headache, and other symptoms of increased ICP; they recommend following the remaining patients with annual clinical evaluations and ophthalmology examinations. One drawback to the latter approach is that in children younger than 5 years, deterioration becomes apparent only when impairment is far advanced because objective testing of vision is difficult in these young patients. Accordingly, we often obtain a baseline imaging study in children with NF-1 who are younger than 5 years of age, after which time a thorough ophthalmologic evaluation can generally be performed. Subsequent MR imaging studies are performed only in children with newly diagnosed or progressive visual impairment and in those with significant abnormalities on initial MR imaging that do not merit immediate treatment, particularly if the children are still too young to cooperate with a thorough ophthalmologic examination. We no longer routinely image older children and instead prefer annual or semiannual clinical evaluations, which include testing of visual acuity and fields as well as a general physical and neurologic examination, with attention directed toward looking for signs of neuroendocrine impairment. The value of visual evoked potential testing for identifying patients with subtle visual impairment from optic gliomas, who may be most likely to benefit from a detailed imaging examination, remains controversial. However, the aforementioned guidelines do not apply to those children who present with severe visual impairment. In our experience, patients who exhibit significant visual compromise have a high risk for further visual deterioration and require either very close follow-up (e.g., every 3 months) or immediate therapy. These recommendations also do not apply to children who have sizeable, symptomatic lesions on their initial imaging studies because these patients generally require immediate intervention. Symptomatic optic nerve gliomas in patients with NF-1 have in the past been suggested to have a less favorable prognosis for long-term disease control after surgical resection than comparable lesions in patients without NF-149; however, this probably reflects an artifactual inference in studies conducted in the era before MR imaging, given that a lesion that is truly localized to one optic nerve is uncommon in NF-1 and that unilateral treatment of a bilateral process is unlikely to have long-term efficacy. In more recent studies, symptomatic chiasmatic–hypothalamic tumors in patients with NF-1 actually appear to carry a more favorable prognosis for long-term disease control than comparable tumors in patients without NF-1.41,42,44,45,50 For example, Hoffman et al41 noted that whereas only 1 of 23 patients with NF-1 and optic–hypothalamic glioma died of disease progression, 7 of 39 patients without NF-1 died (p = 0.045). Deliganis et al50 also noted that time to progression among children with newly diagnosed symptomatic optic pathway gliomas arising in association with NF-1 was substantially longer than that for patients with sporadic tumors (8.4 years vs. 2.4 years, respectively). After an average follow-up of 10.2 years, only 5 of 16 patients with NF-1 exhibited disease progression.50 In addition, a subset of tumors will exhibit spontaneous regression in the absence of any surgical or adjuvant therapy.51 With increased understanding of the natural history of optic pathway lesions in patients with NF-1, the indications for surgical intervention have narrowed considerably. Because the histologic identity of a given lesion is rarely in doubt, biopsy for purely diagnostic reasons is generally not needed. In the occasional patient with an optic nerve glioma that is clearly unilateral, in whom proptosis and blindness are apparent, surgical resection of the involved nerve from the globe to the chiasm may be considered. Such cases are rare because the majority of patients are found on MR imaging also to have involvement of the chiasm and contralateral optic nerve. In these children, radiotherapy or chemotherapy, both of which are described in detail below, may be preferable. These patients should also be distinguished from the occasional patient with an orbital plexiform neurofibroma that extends backward from the globe toward the anterior cavernous sinus, in whom radical resection of the lesion may be required. Surgery has also been advocated for children with large tumors growing exophytically from the optic chiasm.41,52 However, it remains uncertain whether the long-term results in terms of disease stability and functional outcome that are achieved with aggressive resection represent an improvement over those obtained with nonsurgical approaches.53 In contrast to the tumors described above, for which surgical intervention may have a role, albeit a controversial one, the majority of optic pathway gliomas are clearly not candidates for excision because of their diffuse involvement of the optic apparatus and hypothalamus. Radiation therapy has historically been used in the treatment of these unresectable lesions, and it provides excellent results in terms of disease stabilization and occasionally regression, often leading to significant improvement in visual function.54,55 However, radiation may result in severe cognitive and endocrine deficits56,57 and places the patient at risk for radiation-induced malignancies58 and vasculopathy, such as moyamoya syndrome.59 Accordingly, chemotherapy has come to assume an increasing role in the management of these tumors, particularly in patients younger than 5 to 10 years,44,60,61 in whom the risks for long-term radiotherapy-induced cognitive and endocrine impairment are particularly high and in whom the potential benefits of avoiding, or at least deferring, radiotherapy are substantial. A variety of regimens have been employed, with response rates of 20 to 80% and response or stabilization rates of 75 to 100%.44,60,61 The efficacy of a carboplatin/vincristine regimen in children with low-grade gliomas associated with NF-1 was recently assessed in detail in the Children’s Oncology Group 9952 study, in which the results in terms of disease control appeared to be superior to those in children with low-grade gliomas not related to NF.108 Although children with NF-1 do not appear to differ significantly from those without NF-1 in terms of their response to chemotherapy,60 concerns about their potentially increased risk for secondary leukemias resulting from alkylating agents have provided a rationale for avoiding agents like nitrosoureas and temozolomide in front-line regimens. New molecularly targeted agents directed against NF1-related signaling pathway alterations may provide a preferable future alternative for these tumors. A small percentage of patients with NF-1 develop enlarging lesions within the cerebral (▶ Fig. 48.4a) and cerebellar (▶ Fig. 48.4b) hemispheres that differ in appearance from UBOs. The majority of such lesions are gliomas, which exhibit local mass effect and decreased signal on T1-weighted images with enhancement that may be uniform, ringlike, in the form of a mural nodule, or absent altogether. Most lesions are benign and amenable to complete or nearly complete resection, although a small percentage are malignant. Various intraoperative adjuncts, such as stereotactic guidance, intraoperative imaging, and functional monitoring (described more fully in Chapter ▶ 34) have allowed resection of deep-seated lesions with acceptable morbidity. Fig. 48.4 Magnetic resonance images of (a) deep cerebral and (b) cerebellar enhancing lesions in patients with neurofibromatosis 1. Both lesions were detected on follow-up imaging evaluations after the results of initial studies obtained several years earlier had been negative. The patient depicted in (a) had undergone resection of an optic–hypothalamic glioma 8 years previously. Because these lesions had enlarged progressively, complete resection was undertaken. In both cases, low-grade astrocytoma was detected histopathologically.
48.1 Epidemiology, General Diagnostic Criteria, and Molecular Pathogenesis
48.2 Diagnostic Evaluation
48.2.1 Nonneurologic Manifestations
Neurofibromatosis 1
Neurofibromatosis 2
48.2.2 Neurologic Manifestations
Neurofibromatosis 1
Focal Areas of Increased Signal on T2-Weighted Magnetic Resonance Imaging
Optic Pathway Lesions
Cerebral and Cerebellar Hemispheric Gliomas
Neurofibromatosis 1 and 2
Only gold members can continue reading. Log In or Register to continue


Full access? Get Clinical Tree


