Neurogenetic Basis of Pediatric Neurosurgical Conditions
In the past decade, significant progress has been made in increasing our understanding of the molecular genetic basis of neurosurgical disorders. As an example, an integrative genomics approach to a large cohort of medulloblastomas has shown that these tumors comprise four separate molecular subgroups with distinct demographics, clinical presentations, transcriptional profiles, genetic abnormalities, and clinical outcomes.1 Furthermore, a genome-wide association study has recently identified the first susceptibility gene for moyamoya disease.2 Although the molecular basis of pediatric neurosurgical diseases has not been fully elucidated, the application of advanced techniques in molecular biology to the research of neurosurgical diseases is contributing to our understanding of the molecular basis underlying pediatric neurosurgical conditions. In this chapter, we summarize the genetic basis of neurosurgical conditions like neoplasms, cerebrovascular diseases, and congenital and developmental disorders, especially focusing on common pediatric neurosurgical diseases.
8.1 Neurogenetic Basis of Common Pediatric Neurosurgical Diseases
8.1.1 Neoplasms
Diffuse Astrocytoma
Pediatric astrocytomas are histologically similar to adult astrocytomas; however, the extent to which the molecular pathogenesis of adult astrocytomas is similar to that of pediatric astrocytomas is unclear. Indeed, previous cytogenetic studies of high-grade pediatric astrocytomas in which comparative genomic hybridization (CGH) or fluorescence in situ hybridization (FISH) was used have demonstrated distinct patterns of copy number alterations from adult astrocytomas3 and infrequent copy number alterations in pediatric astrocytomas as opposed to those frequently seen in adult astrocytomas.4,5 More recently, advanced microarray-based genetic approaches have also been used to investigate pediatric astrocytomas. Genome-wide profiling with single-nucleotide polymorphism (SNP) arrays performed in 18 pediatric and 6 adult cases of glioblastoma multiforme (GBM) suggested that pediatric and adult GBMs are two molecularly distinct diseases, even though they have a few copy number alterations in common.6 Similarly, SNP arrays that interrogated copy number alterations in 78 pediatric high-grade gliomas have revealed significant differences in copy number alterations between pediatric and adult GBMs,7 suggesting that pediatric astrocytomas show distinct biological behavior and most likely have a different molecular pathogenesis.
Although amplification of the epidermal growth factor receptor (EGFR) gene is seen in up to 40% of adult glioblastomas and 15% of adult anaplastic astrocytomas, it is not commonly seen in pediatric astrocytomas.8 Nevertheless, high expression levels of EGFR were found by immunohistochemistry in 80% of pediatric non-brainstem high-grade gliomas. Mutations of the PTEN gene at the 10q23 locus are rare in pediatric high-grade astrocytomas,9 but significant correlations between the presence of PTEN mutation and decreased survival have been demonstrated in high-grade pediatric astrocytomas. Moreover, frequent heterozygous deletions of 10q23 have been shown by FISH in 61% of 44 pediatric GBMs, and those cases harboring 10q23 deletions were associated with a significantly shorter overall survival time.10 Mutations of p53 in pediatric high-grade astrocytomas have been reported in fewer than approximately 40% of cases.9 Overexpression of p53 as determined by immunohistochemistry is strongly associated with an adverse outcome in pediatric gliomas.11 In one series of 29 high-grade pediatric astrocytomas, inactivation of the p53/MDM2/p14ARF pathway by mutation of p53, overexpression of MDM2, or deletion of p14ARF was seen in over 95% of the cases, whereas the pRb/cyclin D1/CDK4/p16 pathway was inactivated only in up to 25%, as opposed to over 80% of the corresponding adult tumors.8
Diffuse intrinsic pontine glioma (DIPG) is another entity of diffuse astrocytomas most often seen in the pediatric population; however, relatively little is known about the molecular genetics underlying DIPGs, probably because of a lack of availability of primary surgical specimens. A previous study has found a particularly high rate of p53 mutations (71%) in seven pediatric DIPG cases. Another study has demonstrated EGFR overexpression and/or EGFR amplifications in 14 of 16 high-grade DIPGs, whereas only two of 12 low-grade DIPGs exhibited a low level of EGFR expression.12 More recently, the role of two other genes—namely, PDGFRA and poly (ADP-ribose) polymerase-1 (PARP1)—has been implicated in DIPGs with the use of comprehensive SNP microarrays.7,13
Pilocytic Astrocytoma
Early cytogenetic studies by CGH failed to demonstrate common cytogenetic aberrations in pilocytic astrocytomas, revealing few abnormalities in general. Among the few aberrations, gain of chromosome 7 was one of the most common events seen in pediatric pilocytic astrocytomas. Cytogenetic abnormalities were seen more commonly in patients older than 15 years than in patients younger than 15 years,14 and adult cases had much more complex karyotypes, with multiple areas of gains and losses. One of the most important studies in pilocytic astrocytomas during the past decade was an array-based CGH study that identified gains of the BRAF locus at the 7q34 locus.15 In this study, a tandem duplication of the BRAF locus was seen in 28 cases and p.V600E activating point mutations of BRAF in three cases of 53 pilocytic astrocytomas, demonstrating the potentially important role of BRAF and subsequent activation of the mitogen-activated protein kinase (MAPK) signaling pathway in pilocytic astrocytoma pathogenesis. Further molecular mechanisms with regard to the BRAF gene and MAPK pathway activation have recently been described.16–18 The incidence of p53 mutations in pilocytic astrocytomas is controversial, with some authors reporting only infrequent p53 mutations, whereas other authors report more frequent mutations. Mutations of PTEN have rarely been reported in pilocytic astrocytomas. Protein expression of p16, CDK4, and PTEN was detected in 73%, 61%, and 38% of pilocytic astrocytomas, respectively.19
Medulloblastoma
Medulloblastoma has been reported in the setting of several different hereditary cancer syndromes, including Li-Fraumeni syndrome, Gorlin syndrome, Turcot syndrome, and Rubinstein–Taybi syndrome.20 The Li-Fraumeni syndrome is associated with germline mutations in the p53 gene. Patients with Li-Fraumeni syndrome have an increased risk for the development of embryonal tumors (including medulloblastoma) and choroid plexus carcinoma, among many other cancers. Patients with Gorlin syndrome (nevoid basal cell carcinoma syndrome) have germline mutations in the PTCH1 gene, a component of the sonic hedgehog (SHH) signaling pathway, and up to a 5% risk for developing medulloblastoma. Germline mutations in the SUFU gene, a downstream mediator of the SHH pathway, have also been shown in patients with medulloblastoma.21 Germline mutations of the APC gene, a component of the Wnt signaling pathway, are seen in patients with Turcot syndrome, who have an increased risk for colorectal carcinoma and brain tumors, predominantly medulloblastoma. Children with Rubinstein-Taybi syndrome have a complex developmental disorder that includes broad thumbs and toes, characteristic facies, severe developmental delay, and a predisposition to malignancy, including medulloblastoma, oligodendroglioma, and meningioma. This syndrome is secondary to deletion/mutation of the Crebs binding protein gene (CBP), which functions in three pathways involved in medulloblastoma pathogenesis (SHH, Wnt, and p53).22
The most common cytogenetic changes observed in medulloblastoma are loss of chromosome 17p, often through the formation of an isochromosome 17q (loss of one copy of the short arm and gain of one copy of the long arm) and gain of chromosome 7. Other chromosomes frequently found to be lost are 6, 8p, 9q, 10q, 11 and 16q. Genes frequently amplified and/or overexpressed include MYC, ERBB2, OTX2, PDGFRA, and CDK6.23
Recently, large genomic analyses have shed some light into the genetic landscape of medulloblastoma and allowed its stratification in to molecular subgroups (▶ Fig. 8.1). Four independent studies used integrated genomics and established the existence of a Wnt subgroup, an SHH subgroup, and multiple non–Wnt/SHH subgroups. A recent working committee renamed these last subgroups as group 3 and group 4 medulloblastomas, which are currently still poorly understood.24 The Wnt subgroup has a very good prognosis, with long-term survival rates exceeding 90%. The most common genetic alterations found are CTNNB1 mutations and monosomy 6. SHH medulloblastomas have changes in components of the SHH pathway, such as somatic mutations of PTCH, SMO, and SUFU and amplifications of GLI1 and GLI2. This subgroup is more common in young children and in adults, who have an intermediate prognosis between that of the Wnt subgroup (very good) and that of group 3 (poor). Group 3 is characterized by frequent amplifications of MYC and by changes in genes involved in the photoreceptor or GABAergic (γ-aminobutyric acid) signaling. Patients in this group have a high incidence of metastasis and a dismal prognosis. The most common cytogenetic alterations seen in group 4 medulloblastomas are isochromosome 17q (66%) and loss of the X chromosome (80% of female patients). Genes involved in the neuronal or glutaminergic pathways have also been identified, although their genetic or clinical relevance is yet to be clarified.24
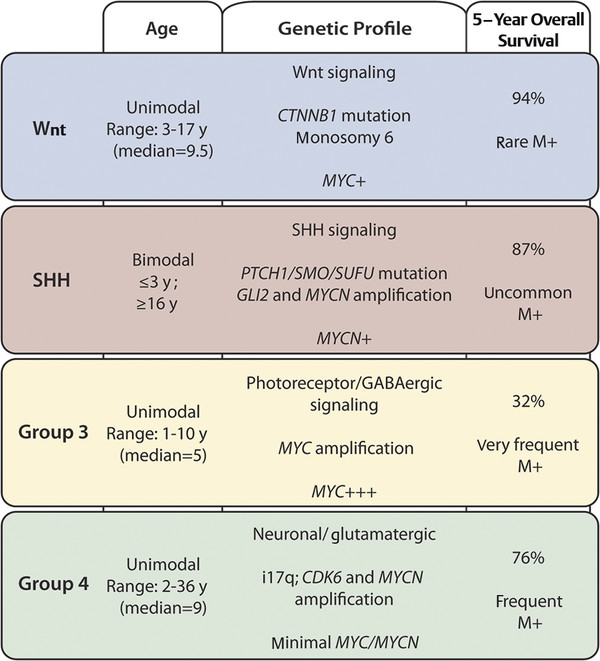
Fig. 8.1 Molecular subgroups of medulloblastoma, including age distribution, genetic profile, survival rate, and incidence of metastasis. GABA, γ-aminobutyric acid.

Fig. 8.2 Molecular subgroups of ependymoma, including clinical and molecular profiles.
Ependymoma
There are few known familial ependymoma syndromes. Spinal intramedullary ependymomas are frequently diagnosed in individuals with neurofibromatosis type 2 (NF2) and are one classic manifestation of the disease. Ependymomas have also been reported in patients with Turcot syndrome (colonic neoplasia and central nervous system neoplasia). These individuals have germline mutations in the APC gene on chromosome 5. Ependymomas have been reported in individuals with multiple endocrine neoplasia type I (MEN1) secondary to mutation of a tumor suppressor gene on chromosome 11q13. Rare families where multiple individuals have ependymomas in the absence of other known germline mutation syndromes have been described.
Over the years, numerous chromosomal abnormalities were reported in ependymoma by studies using karyotyping and CGH. Common genetic anomalies include loss of chromosome 1p, 3, 6q, 9p, 10q, 13q, 16p, 17, 21, and 22q as well as gains of 1q, 4q, 5, 7, 8, 9, 12q, and 20.25 The most common losses seen in children were loss of chromosome 6q, followed by 22q. The regions most frequently amplified in pediatric ependymoma are chromosomes 1q and 9. Gains of 1q are associated with posterior fossa ependymomas, anaplastic features and unfavorable outcome.26
With advances in microarray technology, copy number variations could be mapped at much higher resolutions. Using array CGH, Taylor et al. profiled 103 ependymomas and found three molecularly distinct subgroups, correlated with the anatomical location of the tumor in the supratentorial, posterior fossa, or spinal compartments.27
Similar to array CGH, gene expression profiling studies were able to distinguish tumor-specific signatures according to anatomical location.27,28 Supratentorial ependymomas had high expression levels of EPHB-EPHRIN (EPHB2/3/4 and EPHRINA3/4), NOTCH (JAGGED1/2), and genes involved in cell cycle regulation (CYCLINB2/D1/G2, CDK2/4, and CDKN1C/2C). Posterior fossa tumors highly expressed genes inhibitors of differentiation (ID1/2/4) and members of the aquaporin family (AQP1/3/4). Finally, spinal ependymomas showed up-regulation of various homeobox genes (HOXA7/9, HOXB6/7 and HOXC6/10) and insulin-like growth factor 1 (IGF1). More recently, Witt et al. transcriptionally profiled two large independent cohorts of posterior fossa ependymomas and identified two clinically and molecularly distinct subgroups.29 Group A patients were younger, with tumors located laterally in the cerebellum, and with a balanced genome. This subgroup was also associated with worse clinical outcome and higher incidence of metastasis and recurrence (▶ Fig. 8.2). In contrast, Group B patients had tumors in the midline and a better prognosis. The candidate marker genes to distinguish the two groups were upregulation of LAMA2 in Group A and NELL2 in Group B.29
Other genetic abnormalities were described in ependymomas, including focal amplifications of MYCN, EGFR and YAP1 and deletion of CDKN2A and SULT4A1. The interesting finding from these studies is that up to 40% of pediatric ependymomas appear to have a balanced genomic profile raising the possibility that epigenetic mechanisms may play an important role in ependymoma pathogenesis.
Atypical Teratoid/Rhabdoid Tumors
Atypical teratoid/rhabdoid tumors (AT/RTs) are aggressive embryonic tumors that are usually diagnosed in children younger than 2 years (▶ Fig. 8.3). They are frequently misdiagnosed as medulloblastoma/primitive neuroectodermal tumor (PNET), especially on small biopsies, as up to 70% of rhabdoid tumors contain fields of cells indistinguishable from PNET. As well as occurring in the central nervous system (CNS), rhabdoid tumors are also seen in the kidneys and less commonly in a variety of locations in the soft tissues. Versteege and colleagues identified the tumor suppressor gene on chromosome 22q11 as the SMARCB1 gene, also called hSNF5/INI1.30 The SMARCB1 gene was biallelically inactivated by deletions and/or truncating mutations in a series of renal rhabdoid tumors and cell lines, suggesting that it acts as a classic tumor suppressor gene. This was confirmed by the reintroduction of the gene to cell lines from AT/RTs deficient in SMARCB1, which resulted in dramatic cessation of growth, cell cycle arrest, apoptosis, and expression of markers of cell senescence.31
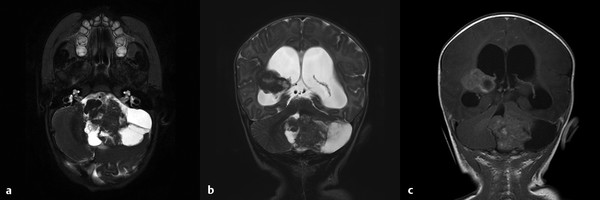
Fig. 8.3 Magnetic resonance images of an atypical teratoid/rhabdoid tumor of the posterior fossa and right lateral ventricle. (a) T2-weighted axial image demonstrating a large mass with cystic component in the posterior fossa and mass effect on the brainstem. (b) T2-weighted and (c) T1-weighted coronal images with gadolinium demonstrating both the posterior fossa and the right lateral ventricle lesions, with obstructive hydrocephalus.
Biegel and co-investigators went on to show somatic deletions and/or truncating mutations of SMARCB1 in CNS rhabdoid tumors.32 It was shown that some children with rhabdoid tumors have de novo germline mutations of SMARCB1, suggesting that these children were born with “one hit” in the SMARCB1 gene and were predisposed to develop rhabdoid tumors.32 In some rare families with an elevated incidence of rhabdoid tumors and/or choroid plexus carcinomas, there may be an inherited germline mutation of the SMARCB1 gene that predisposes affected family members to cancer.20 At least 80% of AT/RTs have genomic mutations of the SMARCB1 gene. Of the remaining 20%, many have decreased expression of SMARCB1 at the protein or RNA level.
The SMARCB1 protein is a member of the SWI/SNF adenosine triphosphate (ATP)–dependent chromatin remodeling complex. It functions to regulate the structure of DNA (chromatin) to either allow or deny access of transcription factors to their respective promoters. The SWI/SNF complex contains about 10 components, raising the possibility that genetic alterations of members other than SMARCB1 could be involved in AT/RT pathogenesis. Recently, inactivation of the ATPase subunit SMARCA4 (also known as BRG1) was shown in the tumor cells of two sisters with rhabdoid tumors lacking the SMARCB1 mutation.33 Another case of a supratentorial AT/RT in a child was reported to have a homozygous SMARCA4 mutation with retained SMARCB1 staining.34
8.1.2 Cerebrovascular Diseases
Cerebral Cavernous Malformation
Cerebral cavernous malformations (CMs), also known as cavernomas, cavernous angiomas, or cavernous hemangiomas, are congenital vascular hamartomas that consist of thin-walled vascular channels without intervening normal brain parenchyma. Although cerebral CMs can occur sporadically, some may arise in an autosomal-dominant inherited form (▶ Fig. 8.4). Genetic linkage analyses in cerebral CMs have mapped and identified cerebral CM genes at three different loci: CCM1 on human chromosome 7q21.2, CCM2 on 7p15-p13, and CCM3 on 3q25.2-q27.35 These cerebral CM genes encode proteins called K-Rev interaction trapped 1 (Krit1), MGC4607, and Programmed Cell Death 10 (PDCD), respectively. The cerebral CM proteins interact with each other and regulate endothelial cell-to-cell adhesion, cell shape remodeling, and cell adhesion to the extracellular matrix, as reviewed recently.36 To date, a number of germline mutations on these genes have been reported, and almost all mutations lead to a premature termination codon, resulting in a loss of protein function. Although the precise mechanism of inherited cerebral CM pathogenesis has not yet been clarified, a two-hit mechanism is implicated in cerebral CM formation,37 as was suggested by Knudson58 in retinoblastoma formation (▶ Fig. 8.5). According to this two-hit model, cerebral CM formation requires a complete loss of any of the three cerebral CM proteins within endothelial cells lining capillary cavities, which results from a germline mutation in one of the two alleles (first hit) and a somatic mutation in the other allele (second hit). In fact, biallelic germline and somatic mutations in each of the three cerebral CM genes have recently been reported, strongly supporting the two-hit mechanism of cerebral CM formation.38–40
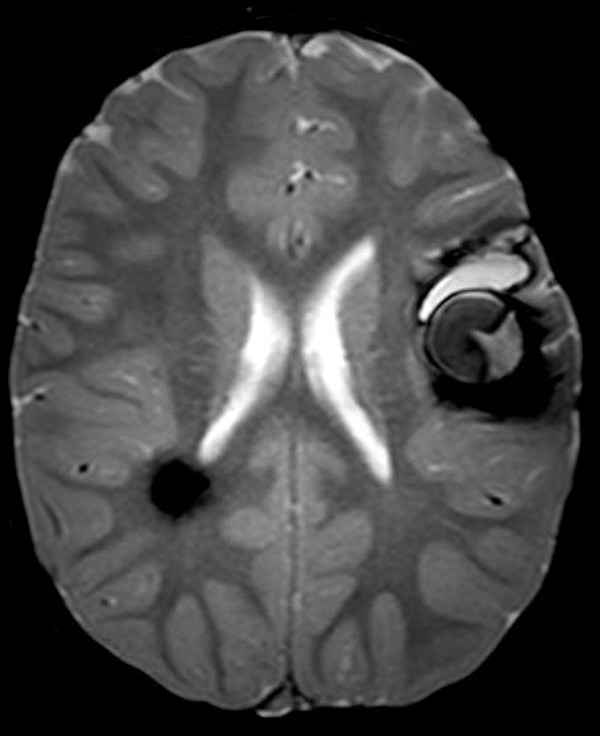
Fig. 8.4 Axial multiplanar gradient-recalled magnetic resonance image showing hemosiderin alterations in the left insular area and right posterior periventricular region. These lesions represent cavernous malformations, and the presence of multiple lesions suggests the diagnosis of familial cerebral cavernous malformation.









