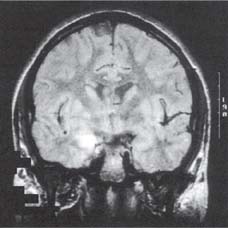
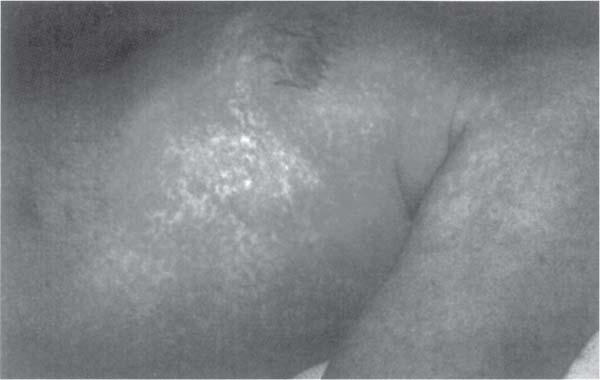
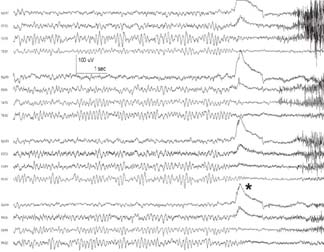
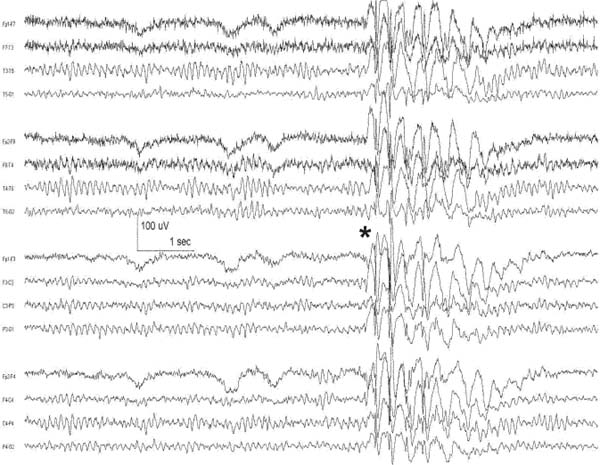
4 Neurology A. The fundamental questions in neurology are 1. Is it neurologic? 2. What is the lesion? 3. Where is the lesion? B. In general, the time course tells you the “what.” 1. Vascular events tend to be spontaneous and rapid in onset. 2. Infectious or metabolic causes tend to evolve over days. 3. Neoplasia and degeneration tend to evolve over months to years. C. Knowledge of functional neuroanatomy/imaging tells you the “where.” 1. Brain, basal ganglia, cerebellum, brainstem, spinal cord, peripheral nerve, neuromuscular junction, muscle A. Developmental milestones (Table 4.1) 1. One year: Single words 2. Two years: Climb 2 steps, 2 word sentences 3. Three years: Tricycle, Repeats 3 digits 4. Four years: Copies a square (4 sides) B. Primitive reflexes 1. Should not persist beyond 3–5 months of age 2. Moro reflex—sudden withdrawal of support results in upper limb (UL) abduction and extension with hand opening followed by UL flexion and adduction; should disappear by 3–4 months; asymmetry or absence suggests focal motor lesion (e.g., brachial plexus injury) 3. Galant reflex—when suspended ventrally, stroking one side of back results in lateral curvature of the trunk toward that side; should disappear at 2–3 months 4. Grasp reflex—should disappear by 2–3 months 5. Tonic neck reflex (“fencing posture”)—disappears by 2–3 months 6. Placing and stepping reflex—disappears by 2–5 months 7. Rooting/sucking disappears by 3–4 months C. Abnormal development 1. Neurocutaneous syndromes a. Neurofibromatosis 1—café-au-lait spots, axillary freckles, Lisch nodules of the iris, neurofibromas, bony lesions; seizures, scoliosis, optic glioma b. Neurofibromatosis 2—bilateral vestibular schwannomas c. Sturge–Weber—port-wine nevus syndrome in V1 distribution, associated vascular malformations of brain with leptomeningeal enhancement, seizures d. Tuberous sclerosis—adenoma sebaceum, ash leaf macules (hypopigmentation), cardiac rhabdomyomas, kidney angioleiomyomas, mental retardation, seizures 2. Motor delay—increased tone with upper motor neuron (UMN) lesion or decreased tone (Werdnig-Hoffman disease [infant spinal muscular atrophy]), Down syndrome, Prader–Willi syndrome (paternally imprinted defect of 15q; Angelman’s if maternally imprinted), basal ganglia, or cerebellar disease 3. Speech delay—male predominance; stuttering affects 2% of children, fluency is increased with ethanol or singing. 90% of lisps and articulation disorders resolve prior to or during adolescence. 4. Cognitive impairment a. Intelligence—the ability to act, think, and deal rationally and effectively with the environment. It is inherited and is located in many parts of the brain. b. Mental retardation—occurs in 3% of births, defined as intelligence quotient (IQ) < 70 c. Learning disability—distinct from mental retardation. Academic difficulties exist in the presence of a normal IQ; rule out mental retardation, dyslexia, seizures, attention deficit disorder, etc. d. Autism—adequate motor and memory skills with decreased social development and communication. Only one third talk; may exhibit stereotypical movements e. Asperger syndrome—autistic but highly functioning in some areas such as math f. Dyslexia—difficulty with written language: poor reading and spelling, but verbal skills are normal. Affects 6% of children, is familial, and is more common in left-handed people. 5. Attention deficit-hyperactivity disorder—3–5% prevalence. Characterized by inattention, forgetfulness, poor impulse control, distractibility. Highly genetic, 50% of cases persist into adulthood. Treatment is with behavioral modification and stimulants such as methylphenidate. Prone to high-risk behaviors and substance abuse (i.e., “self-medication”). D. Aging 1. Changes with aging—decreased pupillary reactions, accommodation, high-tone hearing, taste, smell, strength, reflexes, vibratory sense (with normal proprioception), posture, and gait 2. Dividing cells decrease their rate of division. Nondividing nerve and muscle cells gradually die off. The brain shrinks by 230 g because of neuronal loss. 3. Collagen loses elasticity and contractility. The skull thickens with age, may rarely develop hyperostosis frontalis interna (thickening of frontal bone typically in obese hirsute women), and becomes strongly adhesed to the dura making epidural hematomas much less common than subdural hematomas. 4. Neuritic plaques (first in the hippocampus) and neurofibrillary tangles increase in number. Lipofuscin and iron deposits are increased, as is hippocampal granulovacuolar degeneration. 5. Ataxia develops from cervical spondylosis (spinocerebellar tract damage), cerebellar degeneration, posterior column dysfunction, vestibular degeneration, normal pressure hydrocephalus (NPH), or drugs. A. Memory—involves the dorsomedial thalamus, hippocampus, temporal cortex, ascending reticular activating system (ARAS), and neocortex. Pathways include hippocampus to precommissural fornix to septal gray to diagonal band of Broca to the amygdala. Lesions of the amygdala, fornices, or mamillary bodies do not impair memory. Lesions of the dorsomedial thalamus, hippocampus, or temporal cortex cause memory impairment because these structures are needed for memory. Old memories are stored elsewhere in the brain. B. Acute confusional state—characterized by a decrease in the speed, clarity, and quality of thinking, decreased coherence, orientation, concentration, and recall. There may or may not be illusions, hallucinations, or paranoid delusions. Electroencephalogram (EEG) has high-voltage slow waves. Origin may be drug intoxication, metabolic, concussion, and seizure. C. Delirium—acute, transient, reversible confusion with a disorder of perception, overalertness to stimuli, and increased emotion. Typically autonomic symptoms (unlike other confusional states) such as dilated pupils, tachycardia, hyperthermia, and increased sweating. EEG is normal or has B-waves. Cause must be sought; may result from withdrawal from ethanol, barbiturates, sedatives, herpes simplex virus (HSV), poisons (atropine), respiratory disease, anemia, surgery, trauma, stroke, infection, etc. Delirium tremens has a 10% mortality rate. D. Mood—overall attitude versus affect—outward facial expression in response to a particular stimuli. Mood and affect can be opposite. E. Transient global amnesia—striking loss of memory for recent events and an impaired ability to retain new information, but preservation of remote and working memory. It occurs in middle age or older and typically resolves within 24 hours. Etiology controversial: may be caused by ischemia (thalamus, medial temporal structures), migraine, and temporal lobe seizure; may be functional. Precipitants include physical exertion, overwhelming emotional stress, pain, cold-water exposure, sexual intercourse, and Valsalva maneuver. Psychologic comorbidity is frequent. It has a benign course and seldom recurs. F. Dementia 2. Risk factors include advancing age, positive family history, brain injury, and presence of the apolipoprotein E-4 allele. 3. Important to rule out reversible causes for dementia (10%) including intoxication, infectious, meta-bolic/nutritional, structural, neoplastic, vascular, or mood disorders. “Pseudodementia” results from psychosis or depression. Also consider NPH when there is a triad of dementia, urinary incontinence, and gait disturbance. 4. Alzheimer disease—accounts for over half of dementias with 85% of cases being familial. At age 65, 2–3% prevalence, at age 85, 25–50% have symptoms; progresses to death in 7–10 years. Early short-term memory loss is most striking. Later develops aphasia, apraxia, and agnosia. Characteristic pathologic findings are a. Neuronal loss and atrophy associated with generalized cortical atrophy—more pronounced in temporal and parietal lobes b. Neurofibrillary tangles—paired helical filaments formed by hyperphosphorylation of microtubule-associated protein tau (also seen in Creutzfeldt–Jakob disease and supranuclear palsy) c. Senile plaques – amyloid β peptides, proteolytic product of amyloid precursor protein (APP), average 50 μm in size; when deposited in the walls of small cerebral vessels this causes amyloid (congophilic) angiopathy predisposing to lobar hemorrhage. APP is located on chromosome 21 explaining why those with Down syndrome have symptoms of this disease by age 40. ApoE4 mutation leads to excessive accumulation of amyloid. d. Hirano bodies (intracellular aggregates of actin and associated proteins in neurons also seen in Creutzfeldt–Jakob disease). e. Lowered neurotransmitters, especially acetylcholine (ACh); increased glutamate levels f. Three theories: cholinergic, tau, and amyloid-β hypotheses g. Single photon emission computed tomography (SPECT) is approaching the accuracy of clinical exam (85–90%), although autopsy is required for definitive diagnosis. h. Acetylcholinesterase inhibitors such as donepezil, galantamine, rivastigmine can slow progression, but do not halt or reverse progression. Blockade of N-methyl-d-aspartate receptor excitotoxicity with memantine is showing promise. G. Vascular dementia—second most common dementia, typified by stepwise decline. May be seen concurrently with other forms of dementia. More common in men. Somewhat treatable by treating vascular risk factors. Binswanger disease/subcortical leukoencephalopathy are rare forms of vascular dementia involving deep white matter. H. Pick disease—frontotemporal atrophy; occurs more frequently in young patients; aphasia rare unlike Alzheimer. Socially inappropriate behavior typifies the condition (i.e., inappropriate sexuality, stealing). Genetic causes identified but account for only 5–10% of Pick cases. I. Lewy body disease—10–15% of dementia; overlaps with Alzheimer and Parkinson, typified by α-synuclein cytoplasmic inclusions referred to as Lewy bodies. Loss of both ACh and dopamine-producing neurons. Recurrent visual hallucinations early in the disease along with features of Parkinsonism help establish the diagnosis. J. Wernicke–Korsakoff syndrome—severe, acute deficiency of thiamine (vitamin B1), usually found in chronic alcoholics. Atrophy of mamillary bodies pronounced. Wernicke encephalopathy typically presents with ataxia and nystagmus; Korsakoff psychosis with anterograde and retrograde amnesia and confabulation. A. Cortical structure 1. 2–4 mm thick, forms vertical cortical columns (of Lorente de No), which are functional units 2. Developmentally, inner layers form first with subsequent superficial migration. 3. Three types of cortex: neocortex (90%), paleocortex (base of hemispheres, olfactory system), archicortex (hippocampal formation) 4. Alternate classification a. Allocortex (olfactory cortex and hippocampus)—by definition, variable number of layers b. Isocortex has six layers (refer back to Fig. 1.22): (1) Molecular layer (2) External granular layer (3) External pyramidal layer (4) Internal granular layer (5) Internal pyramidal layer (6) Multiform layer 5. Layer 4 is the chief input layer; layer 5 provides efferent output. Layer 6 is responsible for corticothalamic interconnection. Layers 1, 2, and 3 mediate connections with other parts of the cortex. 6. Pyramidal cells are chief cortical efferents; stellate or granule cells are the main interneurons and are much more numerous. B. Brain zones 1. The central zone (hypothalamus and allocortex)—mediate internal functions 2. The peripheral zone (cortex of sensorimotor and association areas)—mediates perception and interaction with the outside world 3. The border zone (limbic system)—between the central and peripheral zones, adapts the organism to the environment (Fig. 4.1) C. Brain lobes 1. In 1909, Brodmann mapped the cerebral cortex into regions based on cytoarchitecture; this was later found to correlate closely with function. 2. Frontal lobe a. Constitutes about ⅓ of the entire human cortex b. Function—executive functions: personality, motivation, abstract thinking, introspection, and planning c. Components Fig. 4.1 Sensory, motor, and association cortical areas. (2) Premotor (area 6)—contains programming for movements; electrical stimulation produces contralateral movement. Lesion: inability to perform complex movements in the absence of paralysis (apraxia) (3) Supplementary motor (extension of areas 6 and 8, medial frontal lobe)—contains programming for complex movements of several parts of the body. Stimulation results in aversive movements of head, eyes, and contralateral UL. Lesion: transient contralateral weakness, spasticity, and release of suck and grasp reflexes generated by the parietal lobe. Also results in decreased speech initiation that lasts around 3 weeks. (4) Frontal eye field (area 8)—induces contralateral gaze (as in seizure). Lesion: impaired contra-lateral gaze (5) Prefrontal cortex (areas 9, 10, 45, 46)—divided into the orbitofrontal region (visceral and emotional activities) and the dorsolateral area (intellectual abilities and executive functions). It has multiple connections with the visual, auditory, and somatosensory cortices. Stimulation does not elicit a motor response; however, stimulation of the orbitofrontal and cingulate gyri causes autonomic changes such as altered respiratory rate and blood pressure. Lesion: decreased motor activity, compulsive manipulation of objects, and release of some reflexes. Even large lesions here can be asymptomatic. (6) Broca speech (area 44, 45)—pars opercularis, pars triangularis, and pars orbitalis—responsible for the motor component of speech. Lesion: impaired expressive speech distinct from dysarthria, comprehension intact; agraphia and facial apraxia d. Additional frontal lobe symptoms (1) Bilateral frontal lesions—impaired gait and incontinence (lack of warning), pseudobulbar palsy (degeneration of corticobulbar pathways to V, VII, X, XI, and XII cranial nerve nuclei with sparing of III, IV, and VI) (2) Cognitive function—especially with dorsolateral lesions; impaired memory, attention, problem solving, and concentration (3) Initiative—abulia (decrease in thought, movement, speech, will, or initiative) especially with bilateral and anteriorly located lesions. Akinetic mutism is more severe (following eye movements, but no speech or voluntary motor responses), lasts several weeks following lesion. (4) Personality—especially with medial orbital lesions; decreased social consciousness, inappropriate behavior e. Apraxia—the inability to perform a previously learned skill without significant motor, sensory, or ataxic deficits (1) A planned action starts in the dominant parietal cortex and then goes to the premotor and supplemental motor cortices. The parietal area concerned may be near the supramarginal gyrus in the receptive speech center. f. Apraxia types (1) Ideational apraxia—inability to create a plan for a skilled movement. Damage is in the dominant parietal lobe. Test by having the patient act as if he or she is combing the hair without a comb. (2) Ideomotor apraxia—inability to carry out a skilled command. Patient can conceive the movement, but not perform it. Damage is to connections between parietal and frontal lobes. Test by having the patient dress or eat with utensils. Most frequent apraxia. (3) Limb apraxia (4) Limb-kinetic—deficits specifically involving fine limb movements (5) Nonverbal oral/buccofacial—difficulty demonstrating facial movements on command (6) Verbal—impairment of movements necessary for speech (7) Constructional—inability to draw or construct simple structures (8) Oculomotor—impaired eye movement (9) Dressing—impairment in ability to put on clothing g. Motor fibers (1) 30,000 Betz cells in the fifth layer of the motor cortex (area 4), but 1 million axons in each pyramid. The pyramid (in a monkey) contains fibers from the parietal lobe (40%; areas 1, 3, 5, and 7), motor cortex (31%; area 4), and premotor cortex (29%; area 6). Some input also occurs from the supplementary motor area. (2) The corticospinal pathway runs from the motor and sensory cortices diffusely to the intermediate and dorsal horns (nucleus proprius) to control sensory afferent projections and motor function, especially in the face and hands. (3) 80% of the fibers cross, and the uncrossed fibers travel mainly in the ventral corticospinal tract. Most corticospinal fibers synapse on internuncial neurons in the intermediate zone of the spinal gray, and 20% synapse directly on anterior horn cells. (4) Cortical fibers synapse directly on the CN V, VII, nucleus ambiguous, and XII, whereas there is no direct connection to CN III, IV, VI, and dorsal motor nucleus of X. (5) Patients with pseudobulbar palsy are unable to close their eyes or move their mouth or tongue, but are able to yawn, cry, cough, etc., which are reflexes of the pons and medulla. (6) The premotor cortex (area 6) can elicit motor activity with stimulation, but requires a higher amplitude than area 4. (7) Both the premotor and supplementary cortices send fibers directly to the spinal cord. (8) The supplementary motor cortex (anteromedial area 6) elicits gross bilateral movements with stimulation. It receives input from the premotor cortex for planned movements and from the posterior parietal cortex for activity initiated by visual, tactile, and auditory information. Output is to the motor cortex (area 4). (9) A lesion in area 4 produces hypotonia and weakness of the contralateral distal limb, but no spasticity. (10) A lesion in area 6 produces spasticity by increased stretch reflexes. (11) A lesion in the supplemental motor cortex produces involuntary grasping (it normally inhibits this reflexive activity). (12) The ventromedial pathway runs from the tectum, vestibular nuclei, and pontine and medullary reticular nuclei to the internuncial neurons in the ventromedial spinal cord to control axial movements and posture. (13) The lateral pathway runs from the red nucleus to the internuncial neurons in the dorsolateral spinal cord to control the limbs, especially the hands. 3. Parietal lobe a. Functions—body schema; integrating somatosensory, auditory, and visual information. Dominant lobe involved with mathematical calculations and language. Nondominant lobe involved with visuospatial relationships and geographic memory. b. Components (1) Primary somatosensory area (areas 3, 1, 2)—organized like primary motor cortex, size of representation based on sensitivity of a body part rather than size. Stimulation produces contralateral paresthesias. Lesion: loss of contralateral tactile sense and proprioception (3) Primary gustatory cortex (area 43)—anterior portion of parietal operculum. Lesion: contra-lateral ageusia (4) Association area (areas 5, 7)—consists of superior and inferior parietal lobules; processes tactile and visual information; important for awareness of body and environment, performance of sequential tasks, especially if they involve the hands. Stimulation produces no sensation. Lesion: astereognosis, neglect (5) Supramarginal gyrus (area 40)—caps the sylvian fissure (6) Angular gyrus (area 39)—caps the superior temporal sulcus (7) Wernicke area (areas 39, 40, 22)—supramarginal, angular, and posterior portion of the superior temporal gyri c. Additional parietal lobe deficits (1) Vision—deep parietal lesion may interrupt superior geniculocalcarine fibers and produce contralateral inferior quadrantanopsia and also may abolish ipsilateral opticokinetic nystagmus and impair perception of spatial relationships. (2) Cortical sensory functions—lesion may produce astereognosia, graphesthesia, and decreased two-point discrimination, with the deficits bilateral, but more pronounced on the contralateral side. There is decreased localization of touch and pain, but the perception of pain, temperature, touch, and pressure remains intact. (3) Some apraxias can result from parietal lesions—dressing apraxia (mainly nondominant parietal lobe lesion) and constructional apraxia (mainly right side superior parietal lobule) d. Agnosias (“nonknowledge,” or loss of knowledge)—inability to ascribe meaning to stimuli; loss of ability to recognize objects, persons, sounds, shapes, or smells in the absence of sensory deficit or memory loss. Three classifications: visual (occipital lobe), auditory (temporal lobe), tactile (cannot recognize objects placed in hand) e. Gerstmann syndrome—dominant parietal lobe lesion causes right/left dissociation, finger agnosia, acalculia, and agraphia 4. Temporal lobe a. Function—integrates emotion, behavior, and sensation, which merge to create the idea of self. Also plays important roles in hearing, memory, and speech. b. All of the temporal lobe has six layers of cortex (isocortex) except the hippocampus and dentate gyrus, which have three layers (allocortex). c. Vascular supply—medial temporal lobe supplied by the posterior cerebral artery (PCA), superior and lateral temporal lobe supplied by the middle cerebral artery (MCA). d. Anterior and inferomedial temporal lobe have strong connections to the limbic system and are important for visceral activity, emotion, behavior, and memory. Posterior temporal lobe may store experiences; stimulation here produces illusions of past events. e. Left temporal lobe important for learning/memory of verbal information; right temporal lobe is more important for visual information. f. Seizure in the temporal lobe may produce auditory illusions, gustatory sensations, or hallucinations. g. Connections—anterior commissure and middle corpus callosum connect the two temporal lobes. Uncinate fasciculus connects the anterior temporal lobe to the orbitofrontal gyrus. Arcuate fasciculus connects Wernicke to Broca area. h. Components (1) Superior temporal gyrus—involved with acoustic language with output to the limbic and prefrontal areas. Heschl gyrus (areas 41, 42) is the primary auditory cortex located in the posterior superior temporal lobe deep in the sylvian fissure. Stimulation produces humming, buzzing, clicking, or ringing. Lesion: bilateral Heschl gyri injuries may cause deafness. A lesion on one side produces partial loss of hearing on the contralateral side. (2) Areas 22, 21 comprise the auditory association area; stimulation produces sensation of bell or whistle. Lesion: auditory agnosia (3) Middle and inferior temporal gyri—involved with visual discrimination with input from striate and peristriate cortices and output to contralateral visual association cortex, prefrontal, superior temporal, and limbic cortices (4) Vestibular cortex lies just posterior to Heschl gyrus. Lesion: Unilateral lesion results in decreased opticokinetic nystagmus. i. Additional temporal lobe deficits (1) Kluver–Bucy syndrome—from bilateral amygdala damage; absence of emotional response, compulsion to explore all objects visually, tactilely, and orally; hypersexuality and visual agnosia (2) Visual function—interruption of the inferior geniculocalcarine fibers (Meyer loop) produces “pie in the sky” contralateral superior quadrantanopsia. Bilateral lesions of middle and inferior temporal gyri may produce psychic blindness (one can see the object, but not understand what it is or what it is for). Temporal seizures may produce visual hallucinations. (3) Time perception—may be altered by a lesion on either side, a seizure, or Korsakoff psychosis (4) Smell and taste—Smell is affected by a lesion in the posterior orbitofrontal, subcallosal, anterior temporal, or insular cortex. Taste is affected by a lesion in the posterior insula. Olfaction plays a critical role in taste, as in a CN I deficit, commonly seen following brain injury. j. Auditory agnosia—occurs with a lesion in association areas 22 and 21; sounds and tones are discriminated, but there is an inability to recognize words, music, and sounds. It can be separated into auditory agnosia (nonverbal auditory cues) and pure word deafness. 5. Occipital lobe a. Represents one eighth of the cortex. The occipital lobes are connected by the posterior third of the corpus callosum. b. Left side—more involved with naming, symbols, and color Right side—more involved with spatial relationships, forms, faces, constructional praxis, and emotion c. Hallucination—the perception of a stimulus that does not exist versus illusion—the distorted perception of an existing stimulus d. Components (1) Primary visual cortex (area 17) also known as the striate area and receives the optic radiations. Band of Gennari divides the fourth layer of the cortex into two granular layers with a thick myelin layer. Macular vision is represented in the posterior half of area 17. (2) Parastriate cortex (area 18) and peristriate cortex (area 19) have no line of Gennari. These receive input from area 17 and perform complex visual processing (i.e., color, movement direction) e. Deficits (1) Cortical blindness—usually from bilateral PCA strokes and associated with the absence of α waves on electroencephalogram (EEG) (Fig. 4.2) (2) Anton syndrome—an anosognosia; cortical blindness with an affected association area such that the patient denies that he or she is blind (3) Impaired spatial localization—bilateral parietooccipital lesions (4) Balint syndrome—psychic paralysis of gaze with normal extraocular function. Associated with inattention to the peripheral visual field and simultanagnosia; caused by a bilateral lesion in the parietooccipital cortices f. Visual agnosia—inability to recognize objects, lesions of the left occipital and temporal lobes Fig. 4.2 Awake, resting electroencephalogram. Normal posterior α rhythm disappears with eye opening (*). High frequency activity at end of figure after eye opening is muscle artifact. Antero-posterior bipolar montage. (Courtesy of Dr. Richard Wennberg) A. 95% of people are right-hand dominant and have language dominance in the left hemisphere. Of left-handed people, 85% still have language dominance in the left hemisphere, 15% have bilateral dominance, and a very small number have right hemisphere dominance. B. There are two receptive areas— (1) spoken language is received in Heschl’s gyrus (areas 41 and 42) and the posterior superior temporal gyrus (areas 39, 40, 22, and part of Wernicke’s area) and (2) written language is received in the angular gyrus in the inferior parietal lobule (area 39, part of Wernicke area). The supramarginal gyrus lies between these two centers. C. There is one expressive area—the posterior inferior frontal gyrus (area 44, Broca area). D. All speech areas border the sylvian fissure. The arcuate fasciculus connects these areas. E. Aphasias (Fig. 4.3)—evaluate comprehension, reading, speech, writing, repetition, and naming 1. Expressive—lesion in Broca area. Speech output is decreased, with less fluency and impaired repetition (unable to write from dictation but can copy letters). Usually caused by upper branch MCA stroke 2. Receptive—lesion in Wernicke area. Decreased speech comprehension (both written and auditory), fluent par-aphasic speech (using inappropriate, malformed words, or neologisms), impaired reading and repetition, inability to write or say what is wanted. Usually caused by lower branch MCA stroke Fig. 4.3 Classification of aphasia. 3. Global—both Broca and Wernicke areas and is usually caused by large left MCA stroke 4. Conduction—lesion of the arcuate fasciculus. Fluent paraphasic speech, impaired repetition, writing, and reading aloud similar to Wernicke aphasia except with retained understanding of words read and heard. Awareness of the problem preserved. Usually caused by an embolic stroke in the posterior temporal branch of the MCA 5. Transcortical aphasias—lesions at the borderzone of the anterior cerebral artery (ACA), MCA, and PCA; associated with preserved repetition. Transcortical sensory aphasia often associated with hemianopsia and has a good prognosis. 6. Pure word deafness—impaired auditory comprehension and repetition with normal reading, writing, and speaking. Bilateral or unilateral middle third of the superior temporal gyrus between the primary auditory cortex and Wernicke area, often caused by a lower MCA embolic stroke 7. Pure word blindness—alexia without agraphia. Lesion of the left geniculocalcarine tract and corpus callosum A. The amygdala plays an important role in emotion, especially fear; interconnected with olfactory system (afferent) and the hypothalamus (efferent). B. Hunger and thirst are feelings centered more in the hypothalamus. C. Pleasure is concentrated in the nucleus accumbens and septal nuclei. D. Stimulation of the globus pallidus can produce an experience of joy. E. Guilt, anxiety, and paranoia may be associated with the orbitofrontal cortex. F. Anxiety, fear, and depression can be caused by stimulation of the temporal or cingulate gyrus. G. Fear—involves both the sympathetic and parasympathetic systems. Fear is decreased with destruction of the amygdala. H. Anxiety—mainly sympathetic and is associated with depression, hypoglycemia, pheochromocytoma, hyper-thyroidism, and steroids I. Depression—related to low levels of serotonin (5-HT) and norepinephrine (NE) J. Emotional lability—increased control of emotional response with age; may result from cerebral disease K. Pathologic laughing or crying—involuntary and uncontrollable, seen with injury to bilateral corticobulbar tracts from lacunar infarction, multiple sclerosis (MS), and amyotrophic lateral sclerosis (ALS) L. Two paths control the pontomedullary facial movements involved with laughing and crying. The voluntary pathway involves the corticobulbar tract in the genu of the internal capsule. The involuntary pathway is anterior to the genu of the internal capsule. Damage to the anterior path causes unilateral decreased movement with emotion. Damage to the posterior path causes unilateral increased movement with emotion. M. Aggression—may result from birth injury, trauma, encephalitis, or psychomotor seizures. Stimulation of the medial amygdala elicits anger. Bilateral amygdala ablation or section of the stria terminalis reduces anger. N. Placidity or apathy—exploratory behavior is controlled by cortical and limbic dopaminergic pathways to the diencephalon and midbrain through forebrain bundles. Apathy is seen with frontal tumors, NPH, and Alzheimer disease. Abulia or akinetic mutism is caused by bilateral septal nuclei lesions. A. Epilepsy is a chronic condition of various etiologies characterized by a predisposition to recurrent seizures. It is an abnormal and excessive discharge of brain neurons with hypersynchrony and behavioral change. B. 1% of the population. 8% of the population will have a seizure in their lifetime. C. Epileptics have 2–4 times the mortality rate as compared with normal, highest in the first 10 years after diagnosis. A single seizure generally does not constitute epilepsy unless accompanied by a cortical lesion or epileptiform anomalies on EEG. D. 70% are well controlled on anticonvulsants; many refractory patients (usually complex partial seizures) are surgical candidates. If a single first-line anticonvulsant fails, all other agents are likely to fail. E. Kindling: Seizures beget seizures. F. Classification 1. Remote symptomatic—due to a known or identifiable brain lesion 2. Cryptogenic—acquired brain lesion that is unknown or not identified 3. Idiopathic—unknown etiology, presumed genetic G. Epilepsy “zones” 1. Irritative zone—area of cortex that generates interictal spikes 2. Ictal onset zone—area of cortex where seizures are generated 3. Epileptogenic lesion—structural abnormality of the brain that is the direct cause of epileptic seizures 4. Symptomatogenic lesion—portion of the brain responsible for initial clinical symptomatology 5. Functional deficit zone—cortical area of nonepileptic dysfunction 6. Epileptogenic zone—area of brain necessary and sufficient for initiating seizures; removal or disconnection required for amelioration of seizures H. Seizure pathophysiology 1. May involve increased excitation (glutamate) or decreased inhibition (i.e., gamma-aminobutyric acid [GABA]), but this certainly oversimplifies the situation. 2. Some have hypothesized “sick neuron” theory where a neuron becomes hyperexcitable or bursts when it usually would not. 3. Many genetic anomalies are being identified; often involve Ca2+, K+, or Na+ channels or neurotransmitter receptors. 4. Neural synchrony may result from electrotonic interactions via neuronal/glial gap junctions, field effects (lamellar organization of cortex and limbic structures allows for the generation of large electric fields). 6. Depolarization spreads until the surrounding neurons inhibit it. Seizures start in the cortex, spread to the deep nuclei, and then return to the cortex. The impulses go to the basal ganglia, thalamus, and reticular formation where they are amplified (tonic phase, decreased consciousness, autonomic changes, and polyspike pattern). As the diencephalon inhibits the impulse, the pattern changes to clonic with a spike and wave pattern. The depolarizations gradually slow and then stop by the exhaustion of neurons and increased blood–brain barrier breakdown. 7. Todd postictal paralysis may be caused by decreased glucose or increased lactate. 8. Seizure patients demonstrate the frequent loss of hippocampal CA1 neurons and Purkinje cells, possibly caused by hypoxia and ischemia. 9. Febrile seizures—5% of the population has one; as long as they are not complex, do not increase risk of developing epilepsy. Epilepsy risk increases to 6–15% with 2 or more of the following: a. Seizure duration greater than 15 minutes b. Focal seizure c. Abnormal preexisting neurologic exam d. Seizure recurrence within 24 hours e. Family history of epilepsy I. Partial seizures 1. Focal onset; must rule out a structural cause; this is the predominant seizure type after age 40, frequently secondary to stroke 2. Simple (no change of consciousness) or complex (altered consciousness) 3. Also classified as motor, somatosensory, autonomic, or psychic J. Generalized seizures 1. May have bilateral onset or may evolve from a partial seizure 2. May be tonic, clonic, tonic-clonic, myoclonic, or atonic 3. Absence (petit mal) is also a generalized seizure; results from abnormal thalamocortical activity similar to what generates sleep spindles. Seen in 6–12% of epileptics, peak age 4–13 years, and there are frequent automatisms or other clonic activity. The EEG has a characteristic 3 Hz spike and wave appearance (Fig. 4.4). K. Temporal lobe epilepsy (Fig. 4.5) 1. Semiology—may include foul smell (uncus, corticomedial nucleus of the amygdala), Déjà vu (hippocampus), fear and anxiety (central and basolateral nuclei), rising feeling in abdomen (autonomic nuclei) 2. Mesial temporal sclerosis—most frequent cause of intractable temporal lobe epilepsy; typically normal birth history, febrile seizures in 75%; pathology classically notes hippocampal atrophy with gliosis and neural loss in CA1, CA4, and dentate gyrus (Fig. 4.6). L. Pediatric seizure syndromes Fig. 4.4 Absence seizure. Burst of generalized 3 Hz spike and wave activity (*). Primary generalized epilepsy. Antero-posterior bipolar montage. (Courtesy of Dr. Richard Wennberg) Fig. 4.5 Temporal lobe epilepsy. Bilateral temporal lobe interictal epileptiform activity. Independent sharp and slow wave complexes over right (*) and left (**) anterior-midtemporal regions. Anteroposterior bipolar montage. (Courtesy of Dr. Richard Wennberg) 2. Benign epilepsy of childhood with rolandic spikes—focal motor seizures typically involving the face during sleep–wake transition states; conscious but aphasic postictally. Remits spontaneously in adolescence 3. Juvenile myoclonic epilepsy—myoclonus in the morning. Onset is typically 12–16 years; auto-somal dominant condition, variable penetrance; children have normal IQ; 5–10% of cases of epilepsy. Treat with valproic acid for life. 4. Lennox–Gastaut syndrome—multiple seizure types, frequent status epilepticus peak onset 2–6 years. Associated with encephalopathy and brain malformations: children have decreased IQ. Very difficult seizure to treat, frequently proceeded by infantile spasms M. Miscellaneous seizure phenomena 1. Jacksonian march—tonic activity spreading from the fingers, face, followed by lower limbs (LLs), followed by clonic activity 2. Epilepsia partialis continua—persistent focal motor seizures every few seconds. It responds poorly to medications. 3. Reflex epilepsy—induced by auditory, visual, or somatosensory stimuli, or words or eating 4. Neonatal seizures—usually focal. In the first few days, they are ominous and usually related to cerebral damage. In the first few weeks, they are usually due to metabolic disease with decreased glucose, Ca2+, vitamin B6, etc. 5. Pseudoseizures—more frequent in those who have other neurologic conditions. History is key to diagnosis and used to decide who should instead undergo placement of invasive electrodes in the face of an unremarkable EEG. N. Seizure treatment—Because of significant impact of seizures on quality of life, complete seizure freedom is the goal, not a reduction in frequency; may be medical or surgical 1. Anticonvulsants—can be teratogenic (4% birth defects versus 2% in general population, especially cleft lip or palate), but are less damaging than active seizures during pregnancy. Three modes of action, some drugs have multiple modes of action: a. Voltage-dependent Na+ channel blockade Fig. 4.6 Mesial temporal sclerosis. Coronal proton-density magnetic resonance image with increased signal intensity in the right medial temporal lobe. (From Albright AL, Pollack IF, Adelson PD, Eds. Principles and Practice of Pediatric Neurosurgery. New York, NY: Thieme; 1999. Reprinted by permission.) (1) Phenytoin—hold channels longer in their inactive state; allows neurons to fire at moderate, but not very rapid rates. Effective against many seizure types. Half-life is 24 hours, and it is not removed by dialysis (Fig. 4.7). Side effects include allergy (fever, rash, polyarthritis), ataxia, diplopia, stupor, hirsutism, gingival hyperplasia, coarse facial features, cerebellar degeneration, peripheral neuropathy, and decreased vitamin K (supplement pregnant women before delivery). Can cause Stevens–Johnson syndrome. Avoid using with Coumadin (Bristol-Myers Squibb, New York, NY), sulfa, disulfiram, and chloramphenicol. Serum level is increased by cimetidine, chloramphenicol, valproic acid, and uremia. Fig. 4.7 Dilantin rash characterized by maculopapular trunk and proximal limb rash. (2) Carbamazepine—similar mechanism to phenytoin. Half-life is 12 hours. Side effects include leukopenia, pancytopenia (monitor blood counts), diplopia, and hyponatremia from the syndrome of inappropriate antidiuretic hormone (SIADH). Serum levels increased with erythromycin. b. Enhancement of GABA-A system (1) GABA channels mediate inward Cl− current, which hyperpolarizes neurons making them less excitable. Includes barbiturates (which can directly activate these channels) and benzodiazepines (increase channel activity, but cannot activate them). Valproic acid acts by increasing GABA activity. It is hepatotoxic if < 2 years; serum levels are increased with phenytoin (Dilantin, Pfizer Pharmaceuticals, New York, NY) and phenobarbital. Half-life is 8 hours. c. Bind to L-type Ca2+ channels (1) Some anticonvulsants are thought to bind to L-type voltage-gated Ca2+ channels, which are particularly important in the thalamus and thought to be important in absence seizures. Ethosuximide is such a drug (side effects: lethargy, hiccoughs, rare eosinophilia, leukopenia). 2. Other anticonvulsant agents a. Acetazolamide—direct inhibition of carbonic anhydrase or due to resulting acidosis, may be useful in absence and nonfocal epilepsies. Side effects: teratogenic, risk of sulfonamide allergy b. Gabapentin—does not interact with GABA receptors. May be used for generalized or partial seizures, but not absence seizures. Side effects: few side effects include somnolence, dizziness, ataxia, fatigue, nystagmus, increased appetite d. Felbamate—Used as an adjunct for partial seizures, Lennox–Gastaut syndrome. Side effects: aplastic anemia and hepatic failure; use with caution e. Topiramate—may have activity at Na+ channels, GABA, and glutamate receptors. Adjunct to treating partial seizures. Side effects: cognitive impairment, weight loss, dizziness, ataxia f. Tiagabine—GABA uptake inhibitor 3. Treatment strategy—generalized seizures treated with valproic acid. Ethosuximide has a unique role in absence seizures. Dilantin and carbamazepine are generally used for partial seizures but may also have utility in generalized seizures. Phenobarbital is frequently used in children. 4. Status epilepticus—a medical emergency. Continuous seizure or recurrent seizures without normalization of consciousness for 30 minutes; however, many now consider 10 minutes sufficient for diagnosis. High mortality rate; often require intubation and intensive care management. Subclinical status should be considered in patients who have a depressed level of consciousness not otherwise explained. Typically treat with lorazepam 0.2 mg/kg up to 9 mg. Load with Dilantin—20 mg/kg at maximum rate of 50 mg/min (or 150 mg/min for Fosphenytoin). If patient is already on Dilantin, administer 500 mg. 5. Seizure surgery—Considered for those with disabling, medically refractory seizures for > 1 year. Can perform resections or disconnections. Imaging, EEG, and seizure semiology should be concordant for highest chance of operative success. a. Complex partial seizures—Randomized, controlled trial showed 58% seizure-free in those treated surgically versus 8% in medical group. Extent of hippocampal, parahippocampal, and amygdala resection is a source of debate. Must assess laterality of memory preoperatively (Wada test) to ensure unilateral resection would not be debilitating. Unilateral resection may cure bilateral temporal lobe epilepsy. b. Corpus callosotomy—may be used for atonic seizures (drop attacks) and seizures with secondary generalization; involves division of anterior two thirds; does not stop seizures but prevents bilateral involvement allowing for preservation of consciousness; risks include disconnection syndrome; seizure recurrence can occur with time. c. Lesionectomy for lesional epilepsy—When a lesion seems responsible, removing it is often effective for seizure cure. d. Hemispherectomy—generally accepted that “functional hemispherectomy,” which preserves basal ganglia, has lower risk than “anatomic.” May have a role in intractable infantile seizures. e. Multiple subpial transections—Based on the notion of cortical columns. 5 mm linear incisions can be made for partial seizures emanating from eloquent cortex. A. Multiple sclerosis 1. Autoimmune demyelination affecting the white matter of the central nervous system (CNS) with a predilection for periventricular areas. Lesions in different locations, separated in time, are important for the diagnosis (i.e., a single case of optic neuritis or transverse myelitis may not progress to MS). 2. Prevalence ~50/100,000, more common in temperate zones, female predominance, onset usually in young adulthood. 4. Precise cause remains unclear; abnormal immune response clearly important in disease process, however. Associated with perivascular infiltration of monocytes and lymphocytes pathologically. Both environment and genetics seem important. Abnormal cytokine activity reported (interleukin-12); also associated with HLA-DR2. 5. Elevated immunoglobulin G (IgG) in cerebrospinal fluid (CSF) seen—oligoclonal band pattern 6. Uhthoff phenomenon—optic nerve dysfunction after heat exposure. Neuromyelitis optica (Devic disease) 7. Treatment a. Acute exacerbations treated with intravenous methylprednisolone (speeds recovery, but does not affect degree of recovery). b. Immunomodulatory drugs approved as first line therapies for MS (interferon β-1a, interferon β-1b). Decrease the rate of relapses by one third. c. Traditionally, cyclosporine, azathioprine, and methotrexate have been used to treat progressive disease/prevent relapses. B. Other neuroinflammatory disorders 1. Acute demyelinating encephalomyelitis—considered an isolated postinfectious or postvaccinial attack on the CNS 2. Schilder disease—massive demyelination in children, adolescents with malignant course 3. Baló concentric sclerosis—Disease pattern suggests alternating spared and damaged white matter progressing from the ventricles outward. A. Physiology—Involved in the sequencing and modulation of motor activity by controlling the initiation, amplitude, and velocity of a movement. Also tonically inhibit unwanted movements. The caudate loop is involved with planning and selecting appropriate patterns of movement. When the basal ganglia are dysfunctional, hypokinesia occurs because fewer motor units are recruited and several cycles are needed to produce the intended action. Neurotransmitters are glutamate (from the cortex), ACh (from the caudate and putamen), dopamine (DA, from the substantia nigra pars compacta [SNpc]) and GABA (from the SN pars reticulata [SNpr]). The basal ganglia output is in a constant balance between ACh (positive) and DA (negative). Low levels of DA from the SN allow an increase in the effect of the ACh (positive) from the caudate and putamen on the globus pallidus (GP). B. Symptoms of basal ganglia dysfunction 1. Hypokinesia—reduced number of movements; no deterioration in strength. Examples are decreased blinking, difficulty swallowing saliva, mask facies, and monotone soft speech. 2. Bradykinesia—slow movements 3. Posture—normally controlled by the visual, proprioceptive, and labyrinthine input with motor responses. Basal ganglia disease associated with stooped posture, abnormal righting mechanism. 4. Rigidity—bidirectional increased tension of all muscle groups, most prominent in flexors. No associated change in deep tendon reflexes (DTR). Cogwheel rigidity may be caused by the disinhibition of a tremor associated with basal ganglia disease. 5. Decreased SN input → increased striatal output → decreased medial globus pallidus (GPm) output → increased thalamic and pontine output and rigidity. Therefore, rigidity may be caused by decreased output of the SN or GPm. With this model, the rigidity and bradykinesia of Parkinson disease may be treated by a lesion of the GPm or ventrolateral thalamus (receives input from the GPm) (refer back to Fig. 2.7). 6. Athetosis or hemiballism may result if output from the subthalamus or striatum is decreased because this causes increased GPm output. 7. Dyskinesias—difficulty in or distortion of voluntary movements
I. Introduction
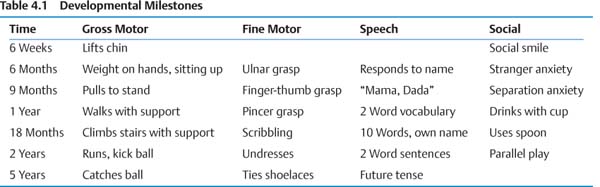
II. Neurologic Development and Aging
III. Confusional States
IV. Brain Structure and Function
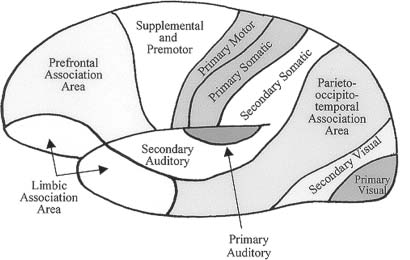
V. Speech and Language
VI. Emotion
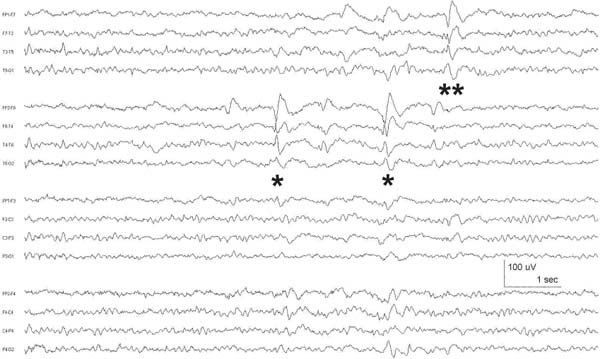
VII. Seizures
VIII. Multiple Sclerosis and Variants
IX. Basal Ganglia
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


