CHAPTER 90 NEUROMUSCULAR JUNCTION DISORDERS
The neuromuscular junction is a prototype synapse but one that is accessible to circulating factors, which makes it a target for neurotoxins and autoantibodies, as well as for genetic disorders. The defects in neuromuscular transmission are associated with well-recognized “myasthenic” symptoms of fatigable weakness, but muscle hyperexcitability syndromes are also recognized. Before the different disorders are discussed, it is important to appreciate the anatomy and function of the neuromuscular junction. The terminal boutons of the motor nerve axon contain mitochondria and small spherical synaptic vesicles that store the neurotransmitter acetylcholine (ACh). Between the presynaptic and postsynaptic membranes is the synaptic cleft, which contains an amorphous-looking basal lamina that, among many functions, anchors the enzyme acetylcholinesterase (AChE) via its collagen tail, ColQ. The peaks of the postsynaptic folds are particularly electron dense because of their high concentration of acetylcholine receptors (AChRs) (Fig. 90-1).
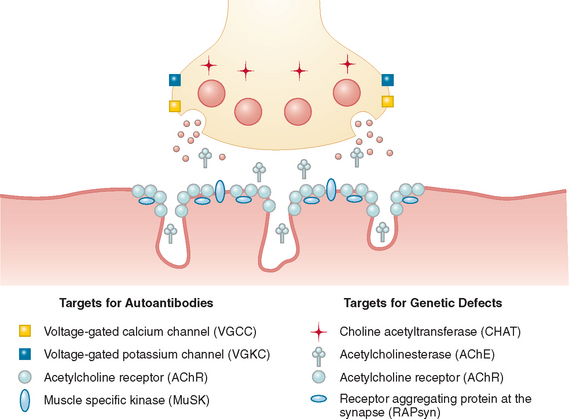
MYASTHENIA GRAVIS
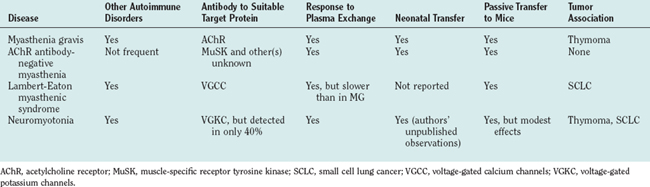
The evidence for autoimmunity at the neuromuscular junction is overwhelming. The observations first made in myasthenia gravis have been extended to other conditions, as summarized in Table 90-1. After observing the thymic pathology in young women with myasthenia gravis, the association with other autoimmune diseases, and the maternal-to-neonatal transfer, John A. Simpson was the first to propose formally that myasthenia gravis was an autoimmune disease. He hypothesized that myasthenia gravis was caused by antibodies directed against an end plate protein. Support for this hypothesis came from two seminal papers published in 1973: (1) a report of reduced AChRs at the neuromuscular junctions of patients with myasthenia gravis1 and (2) the observation that immunization against affinity-purified AChRs could induce AChE inhibitor–sensitive weakness and paralysis in rabbits.2 It remained only to demonstrate antibodies to AChRs in patients,3 as was done by a number of groups over the following years.
Clinical Features and Diagnosis
The weakness often starts in the extraocular muscles, resulting in double vision and ptosis (drooping eyelids), and in 85% of patients, these muscles become involved at some stage. Some patients never develop weakness of other muscles (ocular myasthenia gravis, discussed later), whereas others also develop weakness of bulbar, respiratory, neck, and limb muscles (generalized myasthenia gravis). They complain of difficulty chewing and swallowing, which may result in choking episodes or nasal regurgitation, and their speech becomes slurred, particularly after long conversations. They may develop shortness of breath, especially on exertion and when lying flat (as a result of diaphragmatic weakness). Proximal limb muscles are preferentially affected; hence, the patients have difficulty walking, especially uphill or upstairs, and difficulty holding their arms above their head (e.g., when washing their hair). The characteristic feature of myasthenia gravis that helps to differentiate it from myopathies is the fatigable nature of the weakness. Thus, the weakness varies in distribution and severity from day to day, is often worse at the end of the day, increases with stress, and tends to improve with rest and with use of AChE inhibitors. Table 90-2 lists features that help to differentiate myasthenia gravis from other causes of muscle weakness. Myasthenia gravis was reviewed by Pascuzzi (2004).4
TABLE 90-2 Differential Diagnosis of Myasthenia Gravis
| Symptom | Differential Diagnosis | Distinguishing Feature of Alternative Diagnosis* |
|---|---|---|
| Ptosis and diplopia | Congenital ptosis | Usually obvious from history; if in doubt, ask patient for photos from childhood |
| No associated diplopia | ||
| Mitochondrial cytopathy | Check family history, especially on mother’s side | |
| Diplopia less marked despite extensive limitation of eye movements | ||
| Less fluctuation in symptoms | ||
| There may be other organ involvement | ||
| Oculopharyngeal muscular dystrophy | Family history; genetic diagnosis available | |
| Onset in sixth decade | ||
| Diplopia is rare; ptosis is prominent early, then dysphagia | ||
| Thyroid ophthalmopathy | Look for associated ocular signs, such as proptosis, eyelid lag | |
| Systemic signs and goiter | ||
| Limb muscle weakness | Inflammatory myopathies | Muscles often painful |
| There may be associated features (e.g., rash in dermatomyositis) | ||
| Serum creatine kinase level usually elevated | ||
| Steroid myopathy | History of high-dose daily steroid use | |
| Hip flexors commonly affected | ||
| Muscular dystrophy | Muscle wasting | |
| Family history | ||
| Raised serum creatine kinase level | ||
| Bulbar weakness (especially in elderly patients) | Motor neuron disease | Tongue wasting and fasciculation |
| Look for upper motor neuron signs | ||
| Increased jitter may also be present on single-fiber EMG | ||
| Posterior circulation stroke | Onset is usually more sudden | |
| Patients with less fluctuation usually show steady improvement |
* The feature that differentiates myasthenia gravis from other diseases is the fatigable muscle weakness. EMG, electromyography.
The diagnosis is usually suspected from the characteristic clinical features, especially the fatigability. Confirmatory investigations include detection of AChR or muscle-specific receptor tyrosine kinase (MuSK) antibodies in the serum, electrophysiological testing that reveals a decrement in the compound muscle action potential on 3-Hz stimulation or increased jitter on single-fiber electromyography, and/or a clinical response to short-acting AChE inhibitors (Table 90-3). Rapid improvement after administration of intravenous edrophonium (Tensilon test) is confirmatory in some centers, but objectivity is a problem, and there is a small chance of precipitating cardiorespiratory arrest even if the patient is pretreated with atropine. All patients should undergo thyroid function tests (because thyroid dysfunction is the most common autoimmune disease associated with myasthenia gravis), and imaging of the thorax for detection of an associated thymoma should be performed in patients with AChR antibodies.

Etiology and Pathophysiology
In myasthenia gravis, the numbers of AChRs at the neuromuscular junction are reduced to approximately 20% of normal levels.1 As a result, the EPP is reduced in amplitude and fails to reach threshold in a proportion of muscle fibers, which results in muscle weakness. During repeated effort, the EPP becomes smaller still, and fatigue thus increases. Electron microscopy reveals a normal nerve terminal, loss of the secondary postsynaptic folds, and debris within the synaptic cleft (see Engel5).
About 80% of patients with generalized myasthenia gravis have antibodies to the muscle AChR, which are detected by immunoprecipitation of iodine 125 (125I)–α-Bungarotoxin (α-BuTx)–AChR complexes (α-BuTx is a toxin from Bungarus multicinctus that binds with high affinity and specificity to the AChR). The AChR is a pentameric membrane protein that exists in adult and fetal forms (Fig. 90-2), and α-BuTx binds to each of the two α subunits. The AChR has not been crystallized, but Unwin demonstrated many of the features of the molecule.6
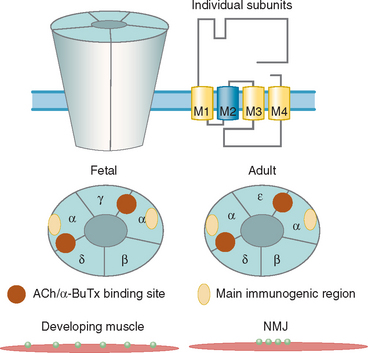
AChR antibodies are high affinity (around 1010 mol), polyclonal, and mainly in the immunoglobulins G1 and G3 subclasses (which are complement-activating). In a proportion of patients, many of the antibodies are directed toward the main immunogenic region on the α subunits7,8 (see Fig. 90-2). Titers of AChR antibody are very variable between patients (ranging from 0 to >1000 nmol) and are not correlated strongly with clinical severity of the disorder. However, the level of antibody within an individual generally changes in parallel with clinical scores after plasma exchange,9 thymectomy,10,11 or immunosuppressive treatments. Moreover, myasthenia gravis can be passively transferred from patients to mice12; the mice may become weak and have reduced numbers of AChRs at the neuromuscular junction.
There are three ways in which the antibodies cause AChR loss in myasthenia gravis. First, the immunoglobulin G (IgG) antibodies can activate complement, causing morphological damage.5 Second, the antibodies, particularly those against the AChR α subunits, can, by binding divalently, crosslink the AchRs, which results in increased internalization and degradation.13 Third, some antibodies can directly inhibit ACh binding to the AChR, which results in direct loss of function.14
Loss of voltage-gated sodium channels, which are located in the secondary folds,15 may increase the threshold for generation of the action potential, thus enhancing the neuromuscular junction defect. However, there is also evidence of a compensatory increase in ACh release16 and of increased AChR synthesis.17 The final distribution and severity of muscle weakness must reflect not only the antibody-induced AChR and secondary sodium channel loss but also these compensatory changes.
Epidemiology
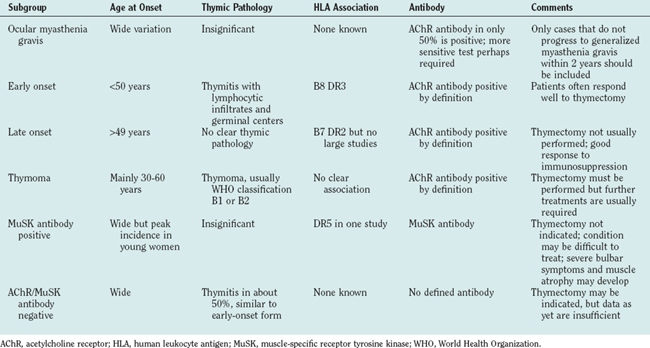
Myasthenia gravis is reported to occur in between 7 and 15 per 100,000 people. These prevalences may be underestimates, inasmuch as current experience suggests that the incidence of new cases may be as high as 1.5 per 100,000 per year,18 and myasthenia gravis remits spontaneously only at the rate of about 1% per year. Patients with myasthenia gravis can be divided into subgroups (Table 90-4), on the basis of age at onset, human leukocyte antigen (HLA) association, thymic involvement, and AChR antibody status.19 However, there may be significant geographical variation; there are few comparative international studies, but in Asian countries more children are affected, and most have ocular myasthenia gravis.
Clinical Heterogeneity
Ocular Myasthenia Gravis
Ocular myasthenia gravis, defined by symptoms that remain restricted to extraocular muscles for at least 2 years, has often been considered a distinct condition (although there may be electrophysiological defects in other muscle groups). AChR antibody levels are generally low or undetectable in about 50% of affected patients. The thymus gland was normal in the few patients examined, and most studies show no apparent HLA association (reviewed by Sommer et al20).
There are differences between ocular muscles and other striated muscles that may determine their selective susceptibility to myasthenic weakness.21 However, ocular weakness is often the presenting symptom in neurotoxin poisoning (e.g., botulism and Bungarus krait envenoming), and it may be that physiological factors or accessibility of the ocular muscle neuromuscular junctions to circulating factors are a major reason for their susceptibility to clinical involvement.
Generalized Myasthenia Gravis with Acetylcholine Receptor Antibodies
Early Onset
There is a 3 : 1 female : male ratio among patients who develop myasthenia gravis in early adulthood, usually defined as younger than 40 years at onset. In Northern Europeans, there is a very strong association with the HLA A1 B8 DRB1*0301 DRB*0101 DQB1*0201 DQA1*0501 ancestral haplotype.22,23
In patients with young-onset myasthenia gravis, the thymus gland is often reported to be “hyperplastic” with many T cell areas in the medulla, some containing lymphoid follicles (for reviews, see Hohlfeld and Wekerle24). Lymphocytes cultured from the thymus gland synthesize AChR antibody,25 and serum AChR antibody levels frequently decline, albeit slowly, after thymectomy.10,11 AChR in its native conformation has been demonstrated in “myoid” cells found in situ between the epithelial cells in thymic medullae both from normal individuals and from patients with myasthenia gravis,26 and these myoid cells may be targets for autoantibody attack, provoking the germinal center formation.27 Affected patients tend to do well with thymectomy and conventional immunosuppressive agents.
Late Onset
Surveys of AChR antibodies have shown a striking increase in incidence after the age of about 60 years, peaking in the late 70s (Fig. 90-3), and even when the declining numbers of the population are taken into account, there is a decrease in frequency of diagnosis of myasthenia gravis after the age of 75 years.18 This suggests that the diagnosis of myasthenia gravis may be missed in elderly persons, partially because of diagnostic confusion with disorders such as motor neuron disease and stroke and partially because of underreporting of symptoms among more elderly patients. In these patients with late-onset disease, the thymus is mainly atrophic or involuted, and there are weak HLA associations. These patients do not have thymomas but nevertheless produce positive results for “thymoma-associated antibodies” to titin, ryanodine receptor, and cytokines.28,29 Thymectomy is seldom performed in patients older than 60 years, and its role in the management of late-onset disease remains unclear. Most patients respond well to low doses of corticosteroids, but often the patients with late-onset disease are slower to respond. They also appear to have a higher incidence of steroid-related complications, such as intestinal perforation.
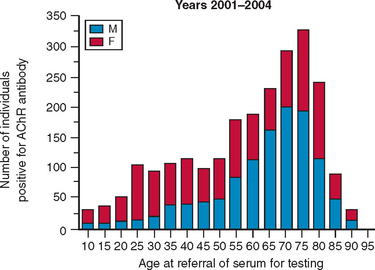
Figure 90-3 Age at referral of 2400 patients with positive acetylcholine receptor (AChR) antibodies as determined by routine diagnostic testing during the period of 2001 to 2004. The samples represent about 50% of the total number of positive AChR results in the United Kingdom. Numbers of male (M) and female (F) patients are approximately equal overall, but women predominate (3:1) among patients younger than 40 years, and men predominate at older ages. It is striking that approximately 75% of the patients present after the age of 40 years; the peak is at approximately 75 years. Statistical analysis suggests that there is a substantial decline in diagnosis of myasthenia gravis after the age of 75 years.18
Thymoma-Associated Myasthenia Gravis
Patients with myasthenia gravis and thymoma can present at any age, but presentation usually occurs between the ages of 30 and 60 years. Thymomas associated with myasthenia gravis are epithelial in origin and correspond mainly to the World Health Organization types B1 and B2 (see Muller-Hermelink and Marx30). More than 90% of patients with thymoma have antibodies to antigens of striated and cardiac muscle,31 including myosin, actin, α-actinin, ryanodine receptor, and titin.32–34 Moreover, antibodies to the cytokines interferon α and interleukin-12, which are found in patients with late-onset disease, are also present in patients with thymoma-related myasthenia gravis and may be predictive of thymoma recurrence.29 Because the thymoma can invade adjacent structures such as the pleura, pericardium, and great vessels and can occasionally metastasize, it should be removed. After removal of the thymoma, however, the myasthenia rarely improves,35,36 AChR antibody levels seldom fall,11 and additional immunosuppressive therapy is usually required. Why the presence of a thymoma leads to myasthenia gravis is not clear, but the gland appears to export large numbers of mature T cells.37






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


