Relative frequency of major molecular pathologic subtypes of frontotemporal lobar degeneration (FTLD). FET, fused in sarcoma, Ewing’s sarcoma, TATA-binding protein-associated factor 15 family of proteins; tau, microtubule-associated protein tau; TDP, transactive response DNA-binding protein.
Normal tau expression and function
Tau is a microtubule-associated protein that binds to and stabilizes microtubules and promotes their polymerization [4]. It is expressed predominantly in axons but also at low levels in glial cells. Tau plays an important role in maintaining neuronal integrity and axonal transport. The MAPT gene is located on chromosome 17q21 and has 16 exons (Figure 13.2A). In the adult human brain, six tau isoforms, ranging from 352 to 441 amino acids, are expressed as a result of alternative splicing of exons 2, 3, and 10 [5, 6]. These isoforms differ from one another by the presence or absence of 29- or 58-amino acid inserts in the N-terminal, and by the presence of either 3 or 4 tandem repeat sequences of 31 or 32 amino acids (3R and 4R tau, respectively) (Figure 13.2A). The repeat regions are the binding domains that mediate the interaction between tau and microtubules. Evidence that tau is more than a microtubule-associated protein comes from observations of other localizations, including the nucleus, plasma membrane, and extracellularly where it interacts with muscarinic receptors. In addition, tau has been found to have a role in cell signaling by interacting with a number of signal transduction proteins [7].
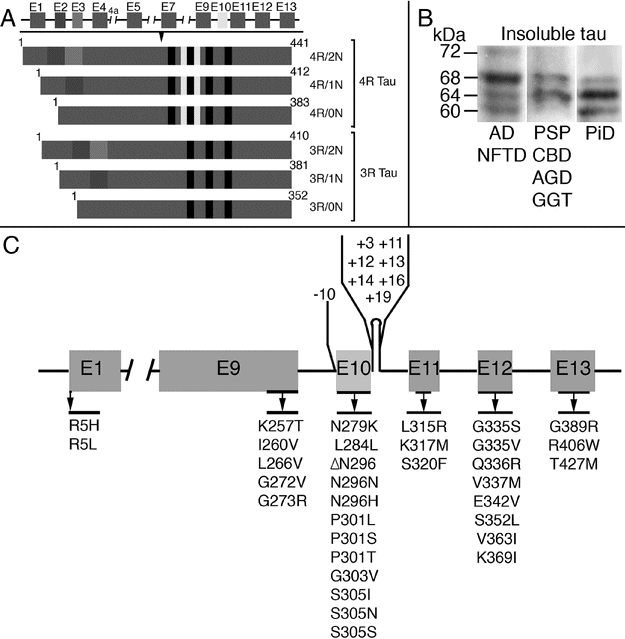
MAPT gene, tau isoforms, and MAPT mutations. (A) Schematic representation of the human MAPT gene and the six tau isoforms. The constitutively spliced exons are E1, E4, E5, E7, E9, E11, E12, and E13. Alternative splicing of exons 2, 3, and 10 give rise to six isoforms (352–441 amino acids). Alternate splicing of exon 10 results in isoforms with either 3 or 4 repeat regions which correspond to the microtubule-binding domains (3R and 4R tau, respectively). (B) Pathologic tau proteins revealed by Western blotting using phospho-dependent tau antibody AT8 in brain tissue from patients affected by different tauopathies. Three different electrophoretic patterns of pathologic tau proteins are illustrated. Pathologic tau bands at 60, 64, 68, and 72 kDa, corresponding to all 6 tau isoforms, are seen in Alzheimer’s disease (AD) and neurofibrillary tangle-only dementia (NFTD). Two major bands at 64 and 68 kDa, representing the 4R tau isoforms, are observed in progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), and globular glial tauopathies (GGT). Prominent bands at 60 and 64 kDa are seen in Pick’s disease (PiD), indicating pathologic tau composed predominantly of 3R isoforms. Cases with MAPT mutations can show all banding patterns, depending on the mutation. (C) Mutations in the MAPT gene in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17 MAPT).
Tau in disease
In the normal adult human brain, similar levels of the 3R and 4R tau isoforms are expressed. Tauopathies are heterogeneous at a biochemical level, with the pathologic form of the protein differing in the relative amounts of 3R versus 4R isoforms [7]. These differences may be demonstrated by immunohistochemistry using 3R- and 4R-specific antibodies and by different banding patterns seen on Western blot analysis of insoluble protein fractions from brain tissue (Figure 13.2B). Diseases in which abnormal forms of both 3R and 4R tau accumulate (e.g., Alzheimer’s disease, AD) have major bands at 60, 64, and 68 kDa, whereas those in which the insoluble protein is composed predominantly of 3R tau only have bands at 60 and 64 kDa and 4R tauopathies only have 64 and 68 kDa bands.
The most studied post-translational modification of tau is phosphorylation, which physiologically regulates its activity and microtubule binding [8]. Tau is normally phosphorylated at 2 or 3 residues, whereas pathologic forms of tau are phosphorylated at 8 to 12 residues, or more. Hyperphosphorylation causes conformational changes and promotes the assembly of tau into abnormal filaments, which adversely affect its interaction with other proteins, and thus has an impact on microtubule stability and axonal transport, dendritic positioning and synaptic health, cell signaling at plasma membranes, protection of DNA from cell stressors, and cellular release of tau [9].
Mutations in the MAPT gene cause autosomal dominantly inherited neurodegenerative disease associated with the intracellular accumulation of soluble and insoluble hyperphosphorylated tau protein (see below). More than 40 mutations have been identified to date (Figure 13.2C) (http://www.molgen.ua.ac.be/FTDMutations/). Mutations either have a primary effect at the protein level or they affect the alternative splicing of tau pre-mRNA [6]. Mutations localized in the microtubule-binding region of tau alter the tau–microtubule interaction and may also have a pro-fibrillogenic effect, while those that affect the alternative splicing of exon 10 lead to an overproduction of 4R isoforms [10].
MAPT is located in a complex genomic region surrounded by three homologous low-copy repeats. A 900 kb genetically balanced inversion in the MAPT genomic region results in two extended haplotypes, H1 and H2 [11]. Inheritance of the H1 haplotype and the H1/H1 genotype is a recognized risk factor for progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) [11]. The H1c sub-haplotype confers additional risk for PSP which may be due to increased tau expression, particularly of the 4R isoforms [12].
Neuropathologic subtypes of FTLD-tau
Pick’s disease
The use of the term has changed over the years, but the designation of Pick’s disease (PiD) is now usually restricted to cases in which the pathology is characterized by classical Pick bodies (see below). The clinical presentation is most often bvFTD or PNFA, with SD being somewhat less common. Rare cases present with an AD-like amnestic syndrome and motor deficits are less common than in most other FTLD subtypes. The typical gross pathology is severe circumscribed frontal and anterior temporal lobar atrophy, which may be asymmetric. Non-specific microscopic features of chronic degeneration, including neuronal loss, gliosis, and swollen neurons (sometimes referred to as Pick cells) are most severe in the frontal and temporal neocortices and limbic cortex, with variable involvement of the striatum and thalamus and relative sparing of the brainstem and cerebellum. The key diagnostic feature is the presence of large, well-demarcated, round or oval neuronal cytoplasmic inclusions that are lightly basophilic, argyrophilic (with Bielschowsky and Bodian but not Gallyas silver stains), and tau-immunoreactive. These Pick bodies are always numerous in the dentate granule cells (Figure 13.3A) and CA1 pyramidal neurons of the hippocampus and are consistently present in affected regions of neocortex [13]. They may also be found in lesser numbers in many subcortical regions. Pick bodies are largely or exclusively composed of 3R tau. Glial tau pathology is less abundant than in other FTLD-tau subtypes, and is represented by ramified astrocytes and tiny globular inclusions in oligodendrocytes.
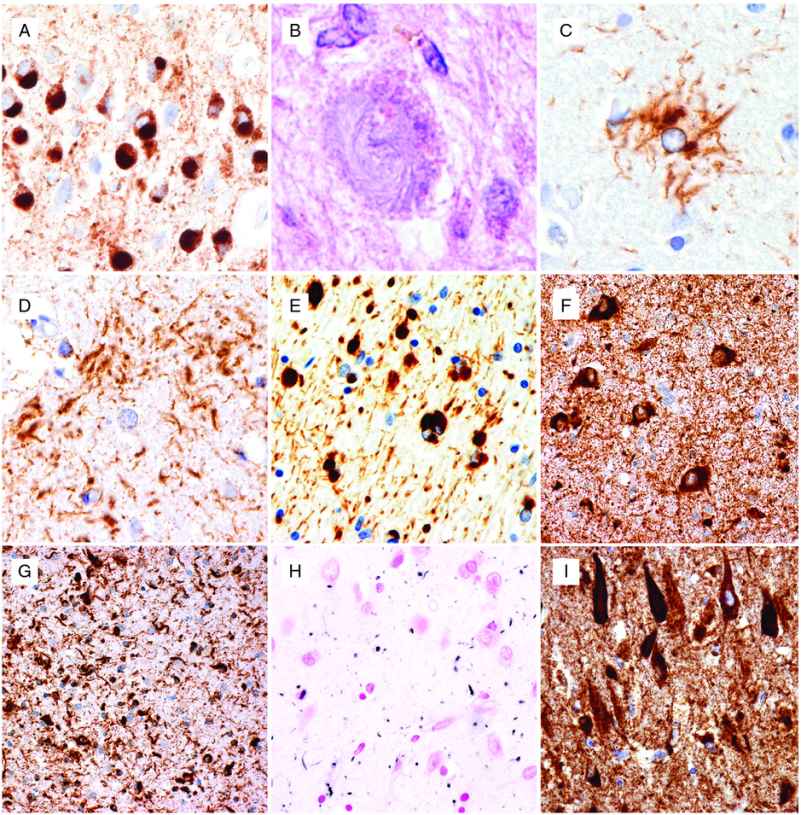
Neuropathologic features of tauopathies. (A) Pick bodies in dentate granule cells of the hippocampus in Pick’s disease. (B) Globose neuronal tangle and (C) tufted astrocyte in progressive supranuclear palsy. (D) Astrocytic plaque in corticobasal degeneration. (E) Globular oligodendroglial inclusions in globular glial tauopathy. (F) Abundant neuronal and (G) glial inclusions in the hippocampus in a case of FTDP-17 MAPT with the S305I mutation. (H) Abundant grains in the hippocampal CA1 region in argyrophilic grain disease. (I) Prominent neurofibrillary degeneration in the hippocampal CA1 region in neurofibrillary tangle-predominant dementia. (A, C–G, I) immunohistochemistry using phospho-dependent tau antibody AT8, (B) hematoxylin and eosin, (H) Gallyas silver stain.
Progressive supranuclear palsy
The classic clinical presentation of PSP, known as Richardson’s syndrome, includes falls, postural instability, axial rigidity, bradykinesia, slow unsteady gait, and ophthalmoplegia. Other presentations, including asymmetric parkinsonism, pure akinesia with gait freezing, CBS, bvFTD, PNFA, apraxia of speech, and primary lateral sclerosis (PLS), are also described [14]. The degree and distribution of gross cerebral atrophy varies and correlates with the predominant clinical features. There is atrophy and discoloration of a variety of subcortical regions, which may include the subthalamic nucleus, globus pallidus, midbrain tectum/tegmentum, substantia nigra, basis pontis, cerebellar dentate nucleus, and cerebellar peduncles. Neuronal and glial cytoplasmic inclusions are best demonstrated with tau immunohistochemistry and Gallyas silver stain. The most characteristic neuronal inclusions are globose neurofibrillary tangles (NFTs) which predominate in subcortical nuclei (Figure 13.3B). Flame-shaped NFTs (similar to AD) may be present in small numbers in the cerebral cortex, and diffuse granular cytoplasmic tau-immunoreactive neuronal “pre-tangles” are present in cortical and subcortical regions. Glial inclusions are present in gray and white matter and include thread-like structures, oligodendroglial coiled bodies, and thorn-shaped astrocytes. However, the most disease-specific pathologic finding is the presence of tufted astrocytes in which tau immunohistochemistry and silver stain highlights a complex branching pattern of proximal and medial cell processes (Figure 13.3C). The neuropathologic diagnosis of PSP is based on demonstrating widespread involvement of subcortical regions by these various neuronal and glial inclusions, all of which are composed primarily of 4R tau. Grumose degeneration of the dentate nucleus of the cerebellum, in which neurons are surrounded by clusters of eosinophilic granular structures that result from the degeneration of presynaptic terminals, is a well-recognized but non-specific feature of PSP [15].
Corticobasal degeneration
The classic clinical presentation of CBD is the corticobasal syndrome, which includes asymmetric ideomotor apraxia, rigidity, dystonia, myoclonus, cortical sensory signs, and alien limb phenomenon. Additional clinical syndromes may include Richardson’s syndrome, posterior cortical atrophy syndrome, bvFTD, and PNFA [16]. Cerebral atrophy is often asymmetric and focal with the distribution correlating with the clinical features. Gross atrophy of subcortical structures is usually not as widespread as in PSP, with the globus pallidus and substantia nigra most often affected. The histopathologic features of CBD overlap with those of PSP and other 4R tauopathies and are best demonstrated with tau immunohistochemistry and Gallyas silver stain. Neuronal inclusions include diffuse granular cytoplasmic tau immunoreactivity, small NFT, globose tangles (sometimes referred to as corticobasal bodies), and occasional small Pick body-like spherical inclusions. Thread-like structures tend to be abundant in the gray and white matter and there are variable numbers of oligodendroglial coiled bodies and thorn-shaped astrocytes. However, the most disease-specific type of inclusion in CBD is the astrocytic plaque, which appears as a circular or ring-shaped collection of short argyrophilic, tau-immunoreactive cell processes that bear a vague resemblance to the neuritic plaque of AD (Figure 13.3D). Unlike the tufted astrocytes of PSP, which involve the more proximal and medial cell processes, astrocytic plaques result from the accumulation of pathologic tau in the distal segments of astrocytic processes, and the associated cell body and nucleus are often not apparent. An additional pathologic feature that was stressed in early descriptions of CBD is the presence of ballooned, achromatic neurons in the cerebral cortex [15]. These are readily apparent on hematoxylin and eosin stained sections and highlighted with immunohistochemistry for neurofilament (NF) proteins and αB-crystallin, but are only weakly argyrophilic and variably tau-positive. Although achromatic neurons may be found in limbic regions in many neurodegenerative conditions, their presence in the neocortex is diagnostically helpful but not specific for CBD.
Globular glial tauopathies
Globular glial tauopathies (GGT) is the term that has recently been recommended for a group of uncommon 4R-predominant tauopathies that are characterized by distinctive and widespread tau-positive globular glial inclusions (GGI) [17]. These cases were previously included in reports of atypical tauopathies under a variety of different terminologies, including “sporadic multiple system tauopathy with dementia.” The clinical presentations include bvFTD with or without extrapyramidal features, PLS, or a combination of FTD and PLS [17]. The distinguishing neuropathologic feature is the globular morphology of cytoplasmic inclusions that occur in both oligodendroglia and astrocytes (Figure 13.3E). While the oligodendroglial inclusions are argyrophilic with Gallyas method, the astrocytic tau deposits are generally silver-negative. Neuronal tau pathology is mainly represented by diffuse cytoplasmic immunoreactivity or globular inclusions, which are also composed primarily of the 4R isoform. Involvement of the white matter is always more prominent than in other tauopathies.
FTD and parkinsonism caused by MAPT mutations
More than 40 different MAPT mutations have been identified in over 100 families with an autosomal dominant inheritance of FTD and parkinsonism (FTDP-17 MAPT), representing approximately 10% of familial FTD cases (http://www.molgen.ua.ac.be/FTDMutations/). Most of the pathogenic mutations are either missense or deletions in exons 1 or 9–13 or mutations in the intron that follows exon 10. The clinical and pathologic features of FTDP-17 MAPT vary significantly, with some degree of correlation with the specific mutation (see below) [18]. The phenotype usually includes some combination of behavior and personality change, cognitive deficits, and atypical parkinsonism. Language deficits, pyramidal dysfunction, and other motor features may also occur but are less common. The neuropathology is characterized by neuronal and glial inclusions (Figure 13.3F–G) composed of hyperphosphorylated, filamentous tau in cortical and subcortical gray and white matter. The anatomical distribution, inclusion morphology, and biochemistry overlap with the sporadic tauopathies. In general, mutations which affect the alternate splicing of exon 10 result in a relative increase in 4R tau and are associated with neuronal and glial pathology that resembles that of sporadic 4R tauopathies (PSP and CBD). In contrast, mutations in exons 9, 11, 12, and 13 result in a predominance of neuronal inclusions; either Pick bodies associated with a 3R-predominant insoluble tau banding pattern or NFT with the same three bands as is found in AD, indicating both 3R and 4R tau isoforms.
Other tauopathies
In addition to atypical and overlapping forms of PSP and CBD, there are reports of sporadic tauopathies that do not fit into current classifications and present as FTD [13, 19]. Various terminologies have been recommended for cases with similar clinical and pathologic features; however, these designations often overlap and are not yet widely accepted. Moreover, comprehensive studies of the aging brain have revealed complex constellations of tau pathologies that involve the frontal and temporal cortices as well as limbic and subcortical structures with various neuronal and glial inclusions, often combined with some AD pathology and even some transactive response DNA-binding protein 43 (TDP-43) proteinopathy [20].
Although argyrophilic grain disease (AGD) and NFT-dementia each rarely present with clinical features of FTD, they are worth mentioning in this context because their pathology often overlaps and may coexist with other forms of FTLD-tau. AGD is a sporadic 4R-predominant tauopathy that usually presents as a late-onset progressive dementia with relatively mild features that are similar to AD [21]. Personality and emotional changes are common but only rarely do patients present as typical bvFTD. Diffuse frontotemporal atrophy is usually mild; however, severe atrophy of the ambient gyrus is frequently documented and it is involved early in disease according to a proposed staging paradigm [22]. The key histopathologic feature is the presence of small dot-like spindle-shaped structures (grains) that are argyrophilic and immunoreactive for tau, ubiquitin, and p62 and thought to represent degenerating dendrites (Figure 13.3H). Grains are most abundant in the amygdala and limbic cortex of the mesial temporal lobe and may extend into the adjacent temporal neocortex. Additional pathologic changes that occur in affected limbic regions include oligodendroglial coiled bodies and silver-negative, tau-positive pre-tangles. Most cases of AGD also have some AD pathology, usually NFT in a Braak stage I–III distribution, and it has been suggested that the presence of argyrophilic grains may lower the threshold for AD-type dementia. A degree of argyrophilic grain pathology has also been reported in a wide variety of other tauopathies (particularly other 4R tauopathies), non-tau-based neurodegenerative diseases, and very old non-demented subjects [21].
A subset of late-onset dementia cases, termed NFT-dementia, is found to have abundant AD-type neurofibrillary pathology (3R- and 4R-positive) that is restricted to the allocortex (Braak stage IV) (Figure 13.3I) with only minimal involvement of the isocortex and without significant numbers of neuritic senile plaques [23]. The clinical presentation is usually relatively mild, slowly progressive dementia that resembles AD and not FTD. Although astrocytic tau pathology is rare, argyrophilic grains and oligodendroglial coiled bodies are often present, suggesting that NFT-dementia and AGD may represent overlapping, age-related conditions.
Models and pathogenic mechanisms
Several basic features of human tauopathies have been recapitulated in model organisms. Experiments in Caenorhabditis elegans have supported the contributory role of tau in neurodegeneration, while those in Drosophila replicate tau hyperphosphorylation, fibril formation, and neurodegeneration. Although these experiments support the notion that the development of tau pathology is linked to clinical symptoms, they also raise the possibility that tau pathology may represent a cytoprotective mechanism aimed at sequestering abnormal tau [7, 24].
Transgenic mice show a variety of phenotypes and pathologic changes that vary with the specific mutations and gene promoters employed. These models include: (1) expression of human wild-type tau isoforms, (2) expression of human mutant tau isoforms, and (3) double and triple transgenic mouse models [24]. The associated pathology includes neuronal tangles, pre-tangles, and glial inclusions. Recent experimental studies that injected extracts from human post-mortem brain with various tauopathies, into the brains of mice transgenic for wild-type human tau, recapitulated many of the hallmark pathologic lesions of the human conditions [25]. Moreover, these experiments demonstrated that once tau inclusions have formed in the brain, they may become self-propagating and spread in a prion-like manner. Taken together, these experimental approaches suggest that tau-mediated neurodegeneration may result from a toxic gain of function of the abnormal tau aggregates, deleterious effects due to the loss of normal tau function, or some combination of both [26].
FTLD-TDP
In 2006, TDP-43 was identified as the pathologic protein accumulating in the vast majority of cases classified as FTLD with ubiquitin-positive inclusions (FTLD-U) and in sporadic amyotrophic lateral sclerosis (ALS) [27]. Accordingly, this pathologic subgroup is now referred to as FTLD-TDP [3]. FTLD-TDP includes sporadic cases as well as familial forms of FTLD with an autosomal dominant pattern of inheritance and clinical symptoms of the FTD spectrum with or without additional features of ALS (Table 13.1). In most series, FTLD-TDP represents the most common pathology underlying FTD (Figure 13.1).
Normal TDP-43 expression and function
TDP-43 is a 414-amino acid protein encoded by the TARDBP gene on chromosome 1, consisting of two RNA recognition motifs, a glycine-rich C-terminal region, and a nuclear localization and nuclear export signal that allows TDP-43 to continuously shuttle between the nucleus and the cytoplasm (Figure 13.4). It is a highly conserved, ubiquitously expressed protein predominantly localized to the nucleus, and expression levels are very tightly regulated by an autoregulatory mechanism. TDP-43 is involved in multiple steps of RNA processing including transcription, splicing, transport, and stabilization [28]. However, the precise functions of TDP-43, particularly in the central nervous system (CNS), are not fully elucidated.

Schematic representation of the TDP-43 protein. GRR, glycine-rich region; RRM, RNA recognition motif; NLS, nuclear localization signal; NES, nuclear export sequence.
TDP-43 in disease
The hallmark lesions of FTLD-TDP are neuronal cytoplasmic inclusions (NCI) and dystrophic neurites (DN) that are immunoreactive for TDP-43, as well as ubiquitin and p62, but negative for other neurodegenerative disease-related proteins, such as tau, α-synuclein, β-amyloid, and fused in sarcoma (FUS) [27]. NCI and DN are characteristically abundant in the frontotemporal neocortex and the dentate granule cells of the hippocampus (Figure 13.5A–G); however, notable differences exist in the morphology and laminar distribution patterns among FTLD-TDP cases, allowing the delineation of four subtypes (see below) (Table 13.2). In a subset of cases, particularly those with a positive family history, neuronal intranuclear inclusions (NII) are also present (Figure 13.5E). Most cases also show TDP-43 pathology in many subcortical regions, including the amygdala, striatum (Figure 13.5H), thalamus, substantia nigra, midbrain tectum and tegmentum, inferior olives, brainstem motor nuclei, and ventral gray matter of the spinal cord (Figure 13.5I). In addition, TDP-43 immunohistochemistry demonstrates previously unrecognized ubiquitin-negative pathology, including diffuse neuronal cytoplasmic staining (“pre-inclusions”) (Figure 13.5J), delicate neurites in the CA1 region in a subset of FTLD-TDP (Figure 13.5K), and glial cytoplasmic inclusions (GCI) in cells of presumed oligodendroglial lineage (Figure 13.5L), that are most abundant in frontotemporal white matter, brainstem, and spinal cord [29].
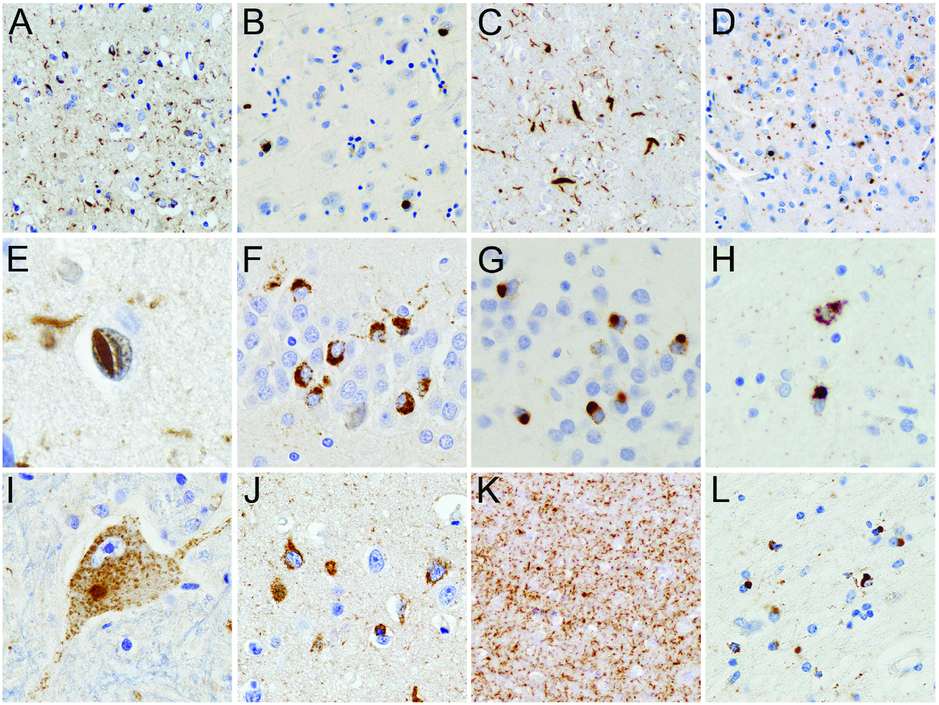
TDP-43-immunoreactive neuropathologic changes in FTLD-TDP. FTLD-TDP is characterized by neuronal cytoplasmic inclusions (NCI), neuronal intranuclear inclusions (NII), and dystrophic neurites (DN) in the frontotemporal neocortex (A–E) and dentate granule cell layer of the hippocampus (F and G) that are immunoreactive for TDP-43. Cases can be subclassified into FTLD-TDP type A in which both NCI and DN are numerous in layer II neocortex (A), FTLD-TDP type B with a predominance of NCI (B), FTLD-TDP type C with a predominance of elongated DN (C), and FTLD-TDP type D characterized by numerous DN and lentiform NII (D and E). Lesser numbers of NII are usually present in FTLD TDP type A cases and are a consistent finding of cases with progranulin gene mutations. NCI in the dentate granule cells of the hippocampus may be granular (F) or compact (G). TDP-43 pathology may be found in many subcortical regions such as the striatum (H) and may be present in lower motor neurons, even in the absence of clinical motor neuron disease (I). Neurons with diffuse granular cytoplasmic reactivity for TDP-43 (pre-inclusions) are most common in cases with type B pathology and a consistent feature in FTD cases with motor neuron disease and those with C9orf72 mutations (J). Numerous delicate DN in the CA1 region of the hippocampus may be associated with severe neuronal loss (hippocampal sclerosis) (K). TDP-43-immunoreactive glial cytoplasmic inclusions are present in cells with oligodendroglial morphology (L). TDP-43 immunohistochemistry.
Neuropathologic subtypes of FTLD-TDP
| Pathologic subtype | ||||
|---|---|---|---|---|
| Neocortical pathologya | Type A | Type B | Type C | Type D |
| NCI | +++ Compact; layer II | ++ Compact, granular, and pre-inclusions; all laminae | + | + |
| DN | +++ Short; layer II | + | +++ Long | +++ Short |
| NII | −/++ | Usually absent | Usually absent | +++ |
| NCI in dentate granule cells of hippocampus | +/++ Granular | +++ Compact and granular | +/+++ Compact | − |
| Subcortical pathology | ++/+++ NCI, DN; basal ganglia, thalamus, brainstem | +/++ NCI; brainstem, spinal cord | ++/+++ NCI, DN; basal ganglia | + NCI, DN, NII; basal ganglia |
a Subtypes are defined by pattern of neocortical pathology.
DN = dystrophic neuritis, NCI = neuronal cytoplasmic inclusions, NII = neuronal intranuclear inclusions.
Semiquantitative grading: − = absent, + = few, ++ = moderate, +++, numerous.
FTLD-TDP is associated with several pathologic changes of TDP-43. The formation of TDP-43-positive inclusion bodies (either cytoplasmic or nuclear) is consistently associated with a dramatic reduction of the normally diffuse nuclear staining (Figure 13.6A) [27]. By immunoblot analysis, a distinct biochemical pattern of TDP-43 is seen in insoluble protein fractions isolated from post-mortem CNS tissue with the presence of FTLD-TDP specific bands at approximately 25 kDa and 45 kDa and a high molecular smear, in addition to the normal 43 kDa band (Figure 13.6B). The pathologic forms of TDP-43 show evidence of abnormal processing with hyperphosphorylation, ubiquitination, and N-terminal truncation [27]. Antibodies raised against phosphorylated epitopes of TDP-43 have further facilitated the detection of pathologic TDP-43 species, since they label only abnormal TDP-43 enriched in inclusions, but not the physiologic TDP-43 (Figure 13.6B–C) [30]. Notable differences are also present in the composition of inclusions with respect to the ratio of full-length TDP-43 versus N-terminally truncated TDP-43 species; while cortical inclusions are selectively enriched for hyperphosphorylated C-terminal TDP-43 fragments, spinal cord inclusions contain more full-length TDP-43 [30, 31].
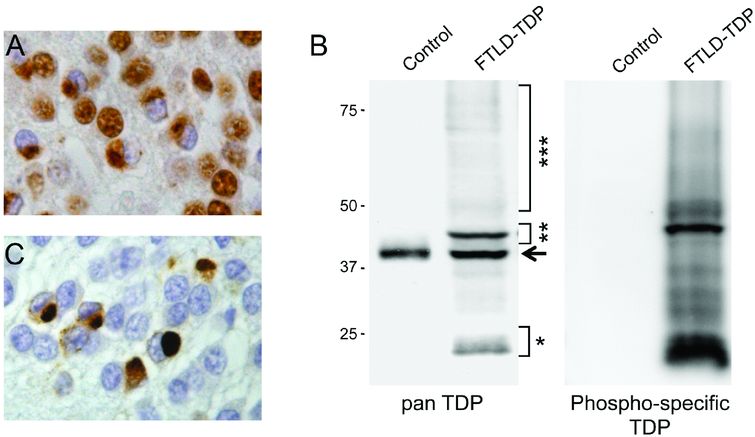
TDP-43 in FTLD-TDP. (A) Cytoplasmic accumulation of TDP-43 is associated with a dramatic decrease of the normal nuclear staining in inclusion-bearing cells, demonstrated with immunohistochemistry using a phosphorylation independent TDP-43 antibody. (B) Immunoblot analysis of urea fractions isolated from brain tissue show the highly characteristic biochemical signature of TDP-43 in FTLD-TDP, with pathologic bands of approximately 25 kDa (*) and 45 kDa (**), and a high-molecular-weight smear (***) that are not detected in controls. The arrow indicates the wild-type 43 kDa TDP-43 band present in controls and in FTLD-TDP patients. With a phosphorylation-specific TDP-43 antibody against phosphorylated serine residues 409 and 410, only pathologic TDP-43 species are detected on immunoblot (B) and in tissue sections (C).
Pathologic subtypes of FTLD-TDP
Prior to the identification of TDP-43 as pathologic protein in FTLD-U, it was noted in two independent studies that FTLD-U cases show a heterogeneous picture with respect to the morphology and laminar distribution of ubiquitin-positive inclusions in the frontotemporal neocortex and three distinct subtypes were delineated (Table 13.2) [32, 33]. Following the identification of TDP-43, and the confirmation that the vast majority of FTLD-U cases have TDP-43 pathology, the same criteria as described in the original ubiquitin immunohistochemistry-based studies were commonly used to subclassify FTLD-TDP cases [34]. Subsequently, a fourth subtype was added, corresponding to the unique pattern of ubiquitin and TDP-43 pathology in VCP mutation carriers [35]. While the findings in both initial typing studies were very similar, different numbering systems were used [32, 33]. To avoid confusion, a harmonized classification scheme was recently introduced, in which the previous numerical subtypes were replaced with subtypes A to D (Table 13.2) [36]. The relevance of the four distinct FTLD-TDP subtypes is supported by relatively specific correlations between the various patterns of pathology, the clinical phenotypes, and the known genetic causes in inherited forms of FTLD-TDP (Table 13.1).
FTLD-TDP type A
Type A cases are characterized by abundant short neuritic profiles and compact oval or crescentic NCI, predominantly in the superficial cortical layers (Table 13.2) (Figure 13.5A). Especially in cases with a positive family history, lentiform NII may be present in affected cortical regions (Figure 13.5E). NCI in the dentate granule cells of the hippocampus are variable in number and often show a granular composition (Figure 13.5F). Few to moderate numbers of GCI are usually present in the cerebral white matter. Frequently affected subcortical regions with TDP-43 pathology are the striatum, thalamus, and midbrain including the substantia nigra.
FTLD-TDP type B
Type B cases show moderate numbers of compact or granular NCI in both superficial and deep cortical layers with relatively few DN and NII (Table 13.2; Figure 13.5B). TDP-43 immunohistochemistry often reveals numerous “pre-inclusions” in cortical and subcortical areas (Figure 13.5J). Characteristically and almost exclusive for type B is the presence of NCI in lower motor neurons, even in the absence of clinical features of ALS (Figure 13.5I). Usually, significant numbers of GCI are present in the cerebral white matter, medulla, and spinal cord.
FTLD-TDP type C
Type C cases have an abundance of long neuritic profiles, predominantly in the superficial cortical laminae, with few or no NCI (Table 13.2; Figure 13.5C). Variable numbers of NCI are present in the hippocampus, usually with a compact, round morphology (Figure 13.5G). NII and GCI are uncommon.
FTLD-TDP type D
The characteristic feature of FTLD-TDP type D pathology is an abundance of lentiform NII and short DN with only rare NCI in neocortical regions (Table 13.2; Figure 13.5D–E) and the absence of NCI in the hippocampal dentate granule cells.
Familial FTLD-TDP
FTLD-TDP due to GRN mutations
In 2006, mutations in the gene encoding progranulin (GRN) were identified in cases of autosomal dominant FTD linked to chromosome 17 that were not due to MAPT mutations [37, 38]. GRN mutations are at least as common in familial FTD as MAPT mutations, accounting for 5–20% of familial FTD and 25–50% of familial FTLD-TDP cases [1]. The clinical presentation is most often bvFTD or PNFA. Extrapyramidal features (akinetic rigid syndrome or CBS) are common but ALS does not occur. Progranulin is a multifunctional secreted growth factor and all pathogenic mutations produce null alleles resulting in a reduction of functional progranulin (haploinsufficiency) [37, 38]. Consistent with this mechanism is the absence of progranulin in the ubiquitin-positive inclusions [37]. Instead, the ubiquitin-positive inclusions in FTLD with GRN mutations are positive for TDP-43 and show a highly consistent pattern of FTLD-TDP type A pathology (Figure 13.5A) [27, 34, 39]. In addition to abundant small NCI and DN in the superficial neocortex, all cases show moderate numbers of lentiform NII (Figure 13.5E). Numerous NCI, DN, and NII are usually seen in the striatum and are more variable in other subcortical regions. In the hippocampus, moderate numbers of TDP-43-positive NCI with a granular appearance are present in the dentate granule cells, and there is often significant loss of pyramidal neurons from the CA1 sector and the subiculum associated with numerous delicate DN (Figure 13.5K).
Related posts:
 Practical management of frontotemporal dementia
The family’s perspective on FTD
Practical management of frontotemporal dementia
The family’s perspective on FTD
 Imaging of frontotemporal dementia
Imaging of frontotemporal dementia
 Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
 Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


