CHAPTER 325 Neuropathology of Traumatic Brain Injury
It is conceptually useful to divide traumatic brain injury (TBI) into primary and secondary types of damage provided that it is understood that the primary types of brain damage are not static but dynamic lesions that evolve over time and thus may be potentially amenable to treatment.1–3
Primary traumatic brain damage is the result of mechanical forces producing tissue deformation at the moment of injury. These deformations may directly damage the blood vessels, neurons and their processes, glia, and microglia in a focal, multifocal, or diffuse pattern and initiate dynamic and evolving processes that differ for each cellular component (Table 325-1). At the least severe end of the spectrum, the changes may be only molecular, and with increasing damage, microscopic and macroscopic lesions become increasingly apparent. This temporal evolution implies that at any given point in time, the pathologic picture is a summative complex of the evolving cascades of damage involving the blood vessels, neurons and their processes, glia, and microglia.4
TABLE 325-1 Features of Primary Traumatic Brain Injury
Note: Most fatal cases of traumatic brain injury are mixtures of focal, multifocal, and diffuse injuries.
Secondary traumatic brain damage (Table 325-2) occurs as a complication of the different types of primary brain damage and includes ischemic and hypoxic damage, expansion of hemorrhagic lesions, cerebral swelling, and consequences of raised intracranial pressure (ICP). Secondary insults such as hypotension, hypoxemia from respiratory complications, electrolyte abnormalities, and pyrexia may further add to the total injury burden.
TABLE 325-2 Features of Secondary Traumatic Brain Injury
2 Focal, multifocal, or diffuse ischemic injury from perfusion failure in microcirculation or macrocirculation |
CPP, cerebral perfusion pressure; ICP, intracranial pressure.
Biomarkers of this structural damage, such as S-100, tau, neuron-specific enolase (NSE), and myelin basic protein (MBP), may have potential utility as diagnostic, prognostic, and therapeutic adjuncts.5
Consciousness is mediated by parallel distributed neuronal networks involving thalamic (cholinergic) and extrathalamic (serotoninergic, noradrenergic, and histaminergic) ascending arousal systems, responsible for wakefulness, and ascending sensory cortical and thalamocorticothalamic loops responsible for awareness of self. These neuronal networks may all be damaged to varying degrees by direct mechanical injury or raised ICP, leading to the various grades of coma, or may be differentially damaged, as in the vegetative state in which the circuits subserving wakefulness are intact but the awareness circuits are not functioning.6
In any given patient, there may be a complex and dynamic interplay of the different primary and secondary types of brain damage and secondary insults to produce a constellation of lesions that is unique both in anatomical site and number. For example, the consequences of primary vascular damage may be bleeding into brain tissue to produce an intracerebral hematoma or interference in the perfusion of the brain tissue with resultant ischemic damage (secondary brain damage), or a combination of the two resulting in increased ICP leading to its sequelae. Thus, head injury is not a single entity but consists of many different types of lesion that may occur rarely in isolation, or more commonly, in varied combinations. This heterogeneity of lesions in TBI makes it unlikely that there is any single pharmacologic agent that will be effective in treating all these intersecting cascades of damage.7,8
Age, genetic predisposition, preexisting disease, drugs, alcohol, and nutritional status are all factors that may influence traumatic injury. The delayed consequences of traumatic injury may continue to evolve for years after the event and include processes such as atrophy, gliosis, neural deafferentation and reinnervation, synaptic plasticity, trans-synaptic degeneration, immune reactions, wallerian degeneration, and neurogenesis.9
Traumatic Axonal Injury
The visualization of damaged axons by traditional silver stains has been greatly improved by a battery of immunocytochemical methods targeting molecules such as amyloid precursor protein-β (APP-β) carried by fast axoplasmic transport and cytoskeletal proteins such as the various neurofilament proteins (NFPs) and tubulins carried by slow axoplasmic transport. Impairment of fast and slow axoplasmic flow leads to progressive axonal swelling and eventual disconnection and formation of axonal retraction bulbs (ARBs) (Fig. 325-1), a process termed secondary axotomy. Current techniques are unable to distinguish traumatic axonal injury (TAI) due to mechanical deformation from axonal injury (AI) due to nontraumatic pathologic processes such as infarcts, hemorrhages, abscesses, neoplasms, and demyelination. The term AI is thus nonspecific and refers to axonal damage of any etiology. TAI may be focal, multifocal, or diffuse. There is increasing recognition of different types of TAI (Table 325-3) and that the anatomic distribution in a detailed neuropathologic work-up including large brain sections may give a clue to the putative mechanism of injury.10
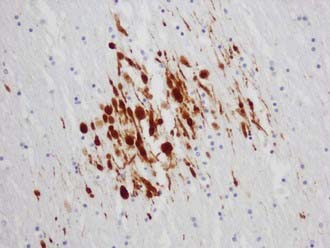
TABLE 325-3 Types of Traumatic Axonal Injury
| Primary axotomy | Tissue tears or lacerations at the severe end of mechanical deformation |
| Secondary axotomy | Progressive impairment of axonal transport resulting in axonal swelling and eventual disconnection with the formation of axonal retraction bulbs |
| Neurofilament compaction | Neurofilament side-arm cleavage |
| Impaired axonal transport without swelling | Impaired axoplasmic transport |
APP-immunopositive axonal damage is an almost universal finding in cases of fatal TBI,11 whereas traditional silver stains only showed damage in about 30% of fatal head injuries.12
Axonal Amyloid Precursor Protein in Traumatic Axonal Injury
APP can be demonstrated immunohistochemically in damaged axons within 35 minutes of the insult.13 APP is normally anterogradely transported along the axon by fast axoplasmic transport as a membrane-bound vesicular protein that accumulates rapidly proximal to the site of injury.14 Reversible APP-β immunoreactive axonal changes have been shown in some experimental animal studies, but whether this also occurs in humans is unknown.
Axonal Amyloid Precursor Protein Patterns in Traumatic Axonal Injury
Multifocal (diffuse) traumatic axonal injury is defined as axonal swellings and bulbs scattered throughout the white matter of cerebral hemispheres, brainstem, and cerebellum as individually affected axons. A spectrum of change is seen that is usually multifocal rather than truly diffuse. Vascular axonal injury (VAI) is defined as axonal swellings and bulbs that cluster around infarct or in ischemic brain in a distribution of vascular compromise associated with raised ICP. The affected axons are often arranged in clusters that have a zigzag, irregular, or geographic pattern15 (Fig. 325-2).
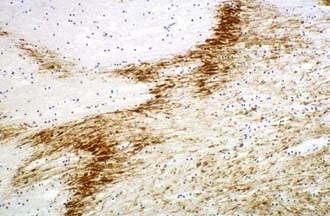
Recent studies in humans have confirmed that APP-immunopositive axons may persist for years after the injury and that this may be associated with the formation of intra-axonal amyloid-β but without evidence of extracellular amyloid-β plaque deposition.16,17 This is in contrast to previous studies that have shown extracellular deposition of amyloid-βcontaining plaque-like structures close to damaged axons just hours after trauma18,19 linking TBI to the development of Alzheimer’s disease.20
Pathogenesis of Traumatic Axonal Injury
Detailed experimental investigation of the pathobiology of TAI has revealed a spectrum of axonal damage.21 Most traumatically injured axons are not mechanically severed at the time of impact as originally believed; instead, they progress gradually to axonal disconnection over several hours.22–24
Axonal deformation at the moment of injury results in a focal impairment of axoplasmic transport and subsequent focal swelling of the axon due to abnormal accumulation of neurofilaments and membranous organelles, and over the next 6 to 12 hours, there is disconnection of the proximal axonal segment from the distal segment. The separated distal segment undergoes wallerian degeneration over time. The transient focal disruption of the axonal membrane allows an influx of Ca2+, which activates multiple Ca2+-dependent deleterious cascades that involve calpains, caspases, and calcineurin and cause disruption of the cytoskeleton. Calcium-linked dephosphorylation of neurofilament side arms results in focal neurofilament compaction (NFC) type of axonal injury.25,26
Diffuse Axonal Injury
Diffuse axonal injury (DAI) was first described as a clinicopathologic syndrome in patients unconscious from the time of trauma, with widespread traumatic axonal damage throughout the brain in the absence of intracranial mass lesions. Similar less severe axonal changes were also found in mild and moderate TBI resulting in the concept that these axonal changes were also the substrate for the transient disorders of consciousness associated with mild and moderate TBI.27 Thus, it was conceptualized that there was a spectrum of DAI, with the severe end of the spectrum correlating with post-traumatic dementia and the mild end of the spectrum correlating with the concussive syndromes.28
The application of more sensitive immunocytochemical techniques such as APP has expanded the spectrum of axonal damage demonstrable in mild, moderate, and severe TBI.29
The severity of DAI has been graded on the basis of the combination of macroscopic and microscopic lesions using silver impregnation techniques to identify axonal swellings and bulbs.12 In grade 1 DAI, widespread axonal damage is present in the corpus callosum, white matter of the cerebral hemispheres, brainstem, and cerebellum. In grade 2 DAI, there are additional focal abnormalities (usually small hemorrhages) in the corpus callosum (Fig. 325-3). In grade 3 DAI, there are, in addition to the findings of grade 2, small focal lesions in the rostral brainstem (Fig. 325-4). Focal lesions in the corpus callosum and dorsolateral rostral brainstem in grades 2 and 3 DAI may be visible on neuroimaging, but in grade 1 DAI without macroscopic focal marker lesions, conventional imaging techniques may not reveal any abnormalities. Neuroimaging of the small focal hemorrhagic lesions in the deep white matter, corpus callosum, and rostral brainstem have been used as surrogate markers of DAI.
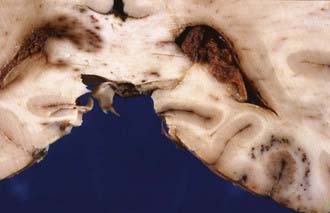

The principal mechanical loading associated with the induction of DAI is rotational acceleration of the unrestricted head, resulting in shear, tensile, and compressive strains that produce dynamic deformation of brain tissue.30 The large size of the human brain plays an important role in the generation of relatively high shear strains between different regions of tissue. The concentration of AI in midline structures may be due to dural barriers such as falx cerebri and tentorium cerebelli, which act as partial barriers to motion of the brain in a given direction.
Although DAI may occur in the absence of impact (contact) forces, most fatal human head injuries are the result of head impacts.31 The contact forces when the head is struck by or strikes a hard object characteristically produce focal lesions such as contusions, but they may also induce rapid acceleration-deceleration, potentially damaging axons. Nonimpact rotational acceleration of the head during car crashes may be followed by single or multiple head impacts against the interior of the motor car or road in the case of pedestrians, motor cyclists, and pedal cyclists.
Traumatic Vascular Injury
There is a large potential spectrum of traumatic vascular injuries that may occur in isolation or in different combinations (Table 325-4). Injury to the intraparenchymal blood vessels may be (1) focal vascular injury, such as contusion, intracerebral hemorrhage, or subarachnoid hemorrhage; (2) multifocal vascular injury, which includes a combination of those injuries; or (3) diffuse vascular injury, such as petechial hemorrhage or microhemorrhage. Injury to the extraparenchymal blood vessels may include (1) injury that bridges veins and arteries, such as acute subdural hematoma (ASDH) or chronic subdural hematoma (CSDH); (2) injury to meningeal arteries and veins, such as extradural (epidural) hematoma (EDH); (3) injury to the venous sinuses, such as EDH; (4) injury to the large arteries in neck; (5) injury to the internal carotid and vertebral arteries, including thrombosis, dissection, subintimal hemorrhage, laceration, and arteriovenous fistula; (6) injury to the blood vessels of the circle of Willis; and (7) injury to the middle, anterior, and posterior cerebral arteries, basilar artery, intracranial internal carotid artery, and vertebral artery, including thrombosis, dissection, subintimal hemorrhage, laceration, and arteriovenous fistula.
TABLE 325-4 Traumatic Vascular Injury
| Intraparenchymal Blood Vessels | |
| Focal vascular injury | Contusions or intracerebral hemorrhage |
| Multifocal vascular injury | Contusions and intracerebral hemorrhage |
| Diffuse vascular injury | Numerous petechial hemorrhages and microhemorrhages |
| Extraparenchymal Blood Vessels | |
| Bridging veins and arteries | Acute subdural hematoma, chronic subdural hematoma, subarachnoid hemorrhage |
| Meningeal arteries and veins | Extradural (epidural) hematoma, subarachnoid hemorrhage |
| Venous sinuses | Extradural (epidural) hematoma, subarachnoid hemorrhage |
| Large Arteries in the Neck | |
| Internal carotid and vertebral arteries | Thrombosis, dissection, subintimal hemorrhage, laceration, A-V fistula |
| Blood Vessels of the Circle of Willis | |
| Middle, anterior, and posterior cerebral arteries; basilar artery; intracranial, internal carotid, and vertebral arteries | Thrombosis, dissection, subintimal hemorrhage, laceration, A-V fistula, subarachnoid hemorrhage |
A-V, arteriovenous.
Cerebral Contusions
Cerebral contusions (bruises) are focal injuries that result when mechanical forces damage the small blood vessels (capillaries, veins, or arteries) and other tissue components (nerve and glial cells and their processes) of the neural parenchyma. The bleeding from damaged blood vessels is usually the most obvious feature on macroscopic and microscopic examination, the manifestations ranging from microhemorrhages to confluent hemorrhage disrupting the tissue (Fig. 325-5). Contusions are typically surface lesions of the brain, but some also include similar hemorrhagic lesions in the deeper structures of the brain.
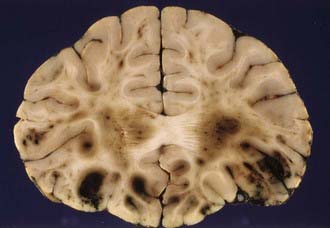
Contusions are dynamic lesions that evolve with time. The progressive expansion or “blossoming” of contusions is demonstrated well by computed tomography (CT) and magnetic resonance (MRI) imaging.32 The damage to the blood vessels sets in train an intertwined cascade of events leading to hemorrhage, breakdown of the blood-brain barrier, and infarction secondary to compromise of the microcirculation (including compromise resulting from thrombotic occlusion of blood vessels).33 This produces a spectrum of macroscopic changes varying from focally dilated blood vessels to burst brain lobes.
Acute surface contusions are characterized by focal vascular damage leading to punctate hemorrhages or small linear hemorrhages aligned at right angles to the cortical surface due to extension of hemorrhage along the perivascular plane. Occasionally, local subarachnoid hemorrhage adjacent to a contusion accumulates within the sulcus to form a sulcal hematoma. This may lead to an erroneous diagnosis of intracerebral hemorrhage on head CT scan. The radiating streak-like cortical hemorrhages on microscopy consist of perivascular accumulations of red cells, and serial sectioning may show evidence of focal traumatic rupture or tearing of the affected blood vessel with bleeding into the perivascular space or neural parenchyma. Damaged blood vessels may thrombose, leading to additional ischemic complications. Contusions often increase in size over hours to days owing to the evolving events related to the interplay of hemorrhage, vasogenic edema, and ischemic necrosis. In the first 24 hours after trauma, contused brain tissue biopsies show an inflammatory response, which is predominantly intravascular and consists of vascular margination of polymorphonuclear leukocytes. Extravascular polymorphonuclear leukocytes can be demonstrated in injured brain tissue only a few minutes after TBI. Three to 5 days later, the inflammation is predominantly parenchymal and consists of monocyte-macrophages, reactive microglia, polymorphonuclear cells, and CD4 and CD8 T-lymphocytes, correlating with delayed postcontusional brain swelling. Inflammatory cells produce free radicals and cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α, which mediate blood-brain barrier injury that leads to brain swelling and induce DNA fragmentation in neurons and oligodendrocytes.34
Surgical and autopsy studies have also provided evidence that apoptosis (an active process requiring energy and protein synthesis) also occurs in human cerebral contusions in addition to necrosis.35 Although necrosis and apoptosis have been considered as distinct separate mechanisms, it is possible that in TBI they represent poles in a continuum of cell injury.36 TUNEL-positive neurons and oligodendrocytes have been identified in human contusions.37 Increased expression of the antiapoptotic protein Bcl-2 has been observed in surviving neurons after human TBI.38 Bcl-2 proteins may participate in the control of cell death and survival by regulating the release of mitochondrial cytochrome c, which is involved in the activation of caspases, especially caspase-3, which cleaves substrates associated with DNA damage and repair, including DNA-fragmentation factor (DFF45/40), poly (ADP-ribose) polymerase (PARP), and the cytoskeletal proteins actin and laminin. Caspase-3 is activated in the injured cerebral cortex of human TBI.38
The next phase is that of resorption of damaged tissue and progressive reactive gliosis. Very small hemorrhages may be completely resorbed within 2 to 3 weeks, whereas larger hemorrhages may take many weeks or months to resorb. The extravasated red blood cells are sequentially broken down to various blood pigments, including hemosiderin. Necrotic brain tissue is phagocytosed by macrophages derived from monocytes at sites where there has been disruption of the blood-brain barrier and lipid macrophages appear 2 to 5 days after the insult. The end result of these processes of resorption is a shrunken brown cystic lesion involving the crests of gyri (plaque jaunt), often with fibrous scarring of adjacent meninges with the formation of a meningocerebral cicatrix (Fig. 325-6).
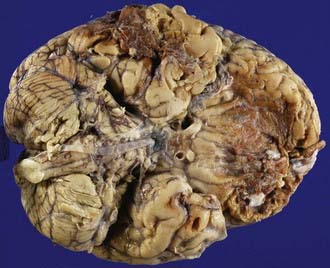
Contusions most frequently involve the inferior frontal lobes and the inferolateral temporal lobes and poles where brain tissue comes in contact with the irregular bony surfaces of the anterior and middle cranial fossae due to the relative motion of the brain and skull at these sites.39 The occipital lobes and cerebellum are rarely contused in the absence of skull fractures.
Types of Contusion
Coup contusions are contusions that occur beneath the site of impact. Coup contusions are a type of contact injury and are produced by compressive forces operating beneath an area of skull in-bending or tensile forces generated by the negative pressure produced beneath an area of skull in-bending suddenly snapping back into place. Contrecoup contusions occur opposite the impact site. Intermediate coup contusions are intracerebral lesions that occur deeply within the neural parenchyma between the impact site and the opposite side of the brain. Fracture contusions occur beneath the site of a fracture. Gliding contusions occur in the parasagittal regions and are believed to be due to the rostrocaudal movement of the brain resulting from impact or impulsive loading forces. The hemorrhages involve the deeper layers of the cortex and the convolutional white matter and spare the surface of the gyrus. Gliding contusions are often associated with DAI.40 Herniation contusions involve the medial temporal lobes and the cerebellar tonsils and are produced by movement of the brain impacting on the rigid tentorium cerebelli or the bony margins of the foramen magnum.
Contusion Patterns and Head Impacts
Previous studies have suggested that the contusion pattern depends on the direction and magnitude of the impacting force and whether the head is accelerated by the impact (e.g., blow to the movable head), is not accelerated by the impact (e.g., blow to the supported head), or is in a state of acceleration at the moment of impact (e.g., fall on head). Thus, a lateral impact in the frontotemporal area may produce a surface contusion of the contralateral temporal lobe and contusions of both uncinate gyri; a lateral temporoparietal impact may result in a contrecoup contusion of the temporal lobe; a midline occipital impact may produce bilateral frontal and temporal lobe contusions; an occipital impact lateral to the midline may cause contrecoup contusions of the frontal and temporal lobes; frontal impacts may result in bilateral or unilateral contusions of the frontal and temporal lobes; and vertex impacts may produce contusions of the brainstem and tears of the corpus callosum and pituitary stalk.41 The surface contusions will be most severe in the frontal and temporal lobes irrespective of the cranial impact site provided the forces acting on the head are sufficient to impart movement of the brain over the irregular bony surfaces of the anterior and middle cranial fossae. Both frontal and occipital impacts result in contusions that are most severe in the frontal lobes, and it therefore cannot be extrapolated that the site of head impact necessarily is diametrically opposite the area of most severe contusion.40
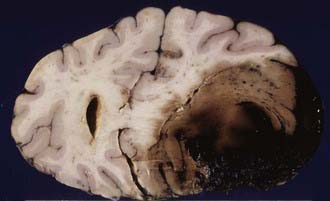
Patients with contusions may show progressive or sudden deterioration. Sudden deterioration is a feature especially of patients with severe bifrontal contusions, temporal pole pulping, and “burst” lobes (Fig. 325-7). Contusions are also one of the causes of neurological deterioration after a lucid interval (“talk-and-die” patients), mimicking extracerebral hematomas.42 However, contusions may be totally absent in patients who have sustained severe or lethal head injury.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


