Fig. 1
Schistosoma mansoni egg surrounded by productive stage granuloma in a spinal cord section. Haematoxylin and eosin stain (courtesy of Prof. E A Bambirra, Universidade Federal de Minas Gerais, Faculdade de Medicina, Departamento de Anatomia Patológica e Medicina Legal, Belo Horizonte, Brazil)
Based on histopathological findings in SCS cases, it has been suggested that ischemic lesions due to immune complex-mediated vasculitis also play a role in the genesis of symptoms in NS (Pittella 1997). Immune complexes containing soluble egg antigen of S. mansoni (SEA) have been demonstrated in the cerebrospinal fluid (CSF) of SCS patients (Ferrari et al. 2011). In this context, investigations of cytokine profiles in CSF and serum of SCS patients suggest the occurrence of an inflammatory and skewed type-2 immune response in the CNS and also systemically (Ferrari et al. 2006; Sousa-Pereira et al. 2006).
The high parasite burden, intense oviposition, and continuous embolization of eggs through collateral vessels of the portal-systemic circulation may explain the higher frequency of egg deposition in ectopic sites, including the CNS, that occurs in association with the most severe chronic forms of schistosomiasis as demonstrated in post-mortem studies (Gonçalves et al. 1995). Although frequent, egg deposition in the CNS of individuals with these severe forms is usually asymptomatic. The lack of symptoms is attributed to the sparse distribution of the eggs and less intense granulomatous reaction that occurs in long-standing schistosomiasis (Pittella 1997).
3.1 Experimental Investigations on Neuroschistosomiasis
Considering the important problems posed by NS, it is surprising that only a few attempts have been made to experimentally study it. Aloe et al. (1996) observed decreased expression of the neurotrophin nerve growth factor in mice infected percutaneously with S. mansoni cercareae, which presented schistosome egg, verified histologically, in the brain. In contrast, Silva et al. (2002) using a similar model, did not succeed in finding S. mansoni eggs in the brain in spite of widespread distribution of eggs in several organs; thus, these authors concluded that the murine model did not appear to be suitable for experimental studies on NS. Recently, Wang et al. (2011) have been able to observe the three stages of the granulomatous reaction and different neurological symptoms, including seizures and motor weakness, after direct injection of S. japonicum egg suspension into rabbit brain. Similarly, neurological symptoms and schistosome egg granuloma in the brain were observed in 90 % of the rabbits which received injection of S. japonicum eggs into their brain (Xu et al. 2013). However, these last two models are different from the natural course of the infection.
Researches aimed at developing a suitable animal model of NS need to be implemented and would be especially useful for the investigation of pathogenetic mechanisms of this disease.
4 Clinical Features, Laboratory Investigations and Diagnosis of Neuroschistosomiasis
All clinical forms of NS may affect men or women at any age, but they are more common in male young adults, teenagers and children.
4.1 Acute Schistosomal Encephalopathy
ASE is more frequent in people without any previous contact with schistosome. Typically, the disease begins about 3 weeks after ASS. The onset of neurological manifestations is usually acute. Headache and some degree of impaired mental status have been observed in virtually all patients. Other common symptoms include focal and generalized seizures, sensory disturbances, weakness of the extremities and cerebellar syndrome. Visual and speech disturbances may also occur. Some of these symptoms may improve and disappear spontaneously in a few days or weeks (Jauréguiberry and Caumes 2008; Ferrari and Moreira 2011).
Computed tomography (CT) and MRI demonstrate multiple focal, small, contrast-enhanced lesions surrounded by oedema. CSF may be normal or may show nonspecific alterations. It is often difficult to detect schistosome eggs in stools or urine, since in recent infection the parasite burden is low. Peripheral marked eosinophilia is a frequent finding. As ASE usually develops in people from non-endemic areas, anti-schistosome antibody detection in serum is useful for the diagnosis. However, these tests may be negative initially. The reliability in detecting early infection by searching for somatic schistosome antigens using monoclonal antibodies remains to be determined (Ross et al. 2007). Polymerase chain reaction (PCR) techniques are being developed for the diagnosis of schistosomiasis with promising results.
In summary, the diagnosis of ASE is largely based on epidemiological and clinical data—especially the history of exposure to contaminated water and presence of ASS a few days or weeks before the neurological symptoms—and positive schistosomal serology.
4.2 Pseudotumoral Encephalic Schistosomiasis
PES typically occurs in people who live in endemic areas, and the patients usually do not have any other manifestation of schistosomiasis. The neurological symptoms develop slowly and are due to tumor-like lesions that cause intracranial hypertension and focal neurological signs, which vary according to the site of the lesion. The pseudotumor may be located in any brain region, but the most common site is the cerebellum, supporting the hypothesis that eggs and/or worms reach the brain through the Batson’s venous plexus (Ferrari et al. 2008b). Extracerebral lesion attached to the dura mater has also been reported (Rommel et al. 2005). PES often presents as a single lesion, but cases of two distinct lesions have been described (Pittella et al. 1996; Roberts et al. 2006).
Headache, seizures, altered mental status, visual abnormalities, speech disturbances, sensory impairment, motor deficits, nystagmus, vertigo, ataxia, nausea, vomiting and papilledema are commonly reported. Focal motor seizures and a cerebral lesion on CT or MRI may be the only evidence of the disease. Symptoms are present from a few weeks to more than 1 year before the diagnosis (Pittella et al. 1996; Ferrari 2004; Betting et al. 2005; Li et al. 2011).
CT and MRI show a nodular, space-occupying lesion with surrounding oedema, and heterogeneous contrast enhancement. T1-weighted MRI may show a central linear enhancement surrounded by multiple enhancing punctate nodules, with an “arborized” appearance (Sanelli et al. 2001). Schistosome eggs may or may not be present in stool or urine. Reports on CSF examination are rare. Peripheral eosinophilia is commonly mild or absent. A positive serum schistosomal antibody test is not sufficient to confirm active disease, as it is only evidence of previous exposure to schistosome antigens. Thus, serology is often devoid of diagnostic value in individuals born and raised in schistosome endemic areas (Ross et al. 2002). Techniques to detect parasite antigens have been developed (Deelder et al. 1994), but they are not yet commercially available.
The diagnose of PES may be difficult because the clinical findings are indistinguishable from those caused by any other tumor-like lesions of slow growth, and laboratory tests are of little help. Therefore, diagnosis of PES relies on the demonstration of eggs and granulomas in nervous tissue biopsy (Ferrari and Moreira 2011).
4.3 Spinal Cord Schistosomiasis
SCS is also more frequent in people who live in endemic areas. The patients usually do not have any other symptom of the infection except for hepatomegaly, which has been observed in about one-fourth of the patients with S. mansoni SCS (Ferrari 2004). There are only a few reported cases during (or soon after) ASS (Pittella 1991) or in association with hepatosplenic schistosomiasis (Ferrari et al. 2001; Araújo et al. 2006).
Typically, SCS presents as a low cord syndrome with acute or subacute progression, usually associated with involvement of the cauda equina roots. The most frequent initial complaint is low back pain, which usually irradiates to the lower limbs, followed by weakness and sensory impairment in the lower limbs, and autonomic dysfunction, particularly bladder dysfunction. Pain intensity varies from mild to very severe, and lower limb weakness can prevent walking in about 63 % of the cases (Ferrari 2004; Ferrari et al. 2008a). Other common symptoms of SCS are lower limbs paresthesia, hypoesthesia or anesthesia, deep tendon reflexes abnormalities, constipation and sexual impotence. The level of the lesion determined by the clinical examination is usually equal or below T6, especially at T11-L1. The low localization in the spinal cord, the acute or subacute progression of symptoms, and the involvement of both the spinal cord and spinal roots are the most characteristic clinical features of SCS (Ferrari 1999).
Atypical presentations of SCS have occasionally been reported and include a slowly progressive form, a waxing and waning myeloradiculopathy that takes months to reach its complete development (Ferrari 1999; Chen et al. 2006; Lighter et al. 2008), and a high thoracic or even cervical localization (Ferrari 1999; Junker et al. 2001).
MRI demonstrates findings of inflammatory myelopathy. The most frequent abnormalities are enlargement of the spinal cord, especially the conus medullaris, and thickening of the cauda equina roots with a heterogeneous pattern of contrast enhancement (Fig. 2). Stool and urine examination fails in revealing eggs in more than half of patients. Rectal biopsy with ovogram allows identification of S. mansoni eggs in 95–100 % of the cases (Ferrari 2004; Silva et al. 2004; Araújo et al. 2006). CSF examination usually shows a slight-to-moderate increase in both total protein concentration and cell count. Mononuclear cells usually predominate and eosinophils may or may not be present. As stated above, a positive serology in serum is virtually devoid of diagnostic value in people from endemic areas. In contrast, because of the segregation of the CNS from the rest of the body due to physiological barriers, the finding of antibodies against schistosome antigens in the CSF has been considered a potentially useful tool in the diagnostic approach to SCS (Pammenter et al. 1991; Ferrari et al. 1995).
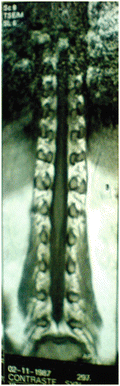
Fig. 2
Coronal gadolinium-enhanced T1-weighted MRI of the spinal cord showing enlargement of the conus medullaris in a patient with spinal cord schistosomiasis due to Schistosoma mansoni
In this context, the measurement of IgG against SEA in the CSF by ELISA for the diagnosis of SCS has demonstrated a high specificity, though with limited sensitivity (Pammenter et al. 1991; Ferrari et al. 1995; Ferrari 2010). Reduction of CSF anti-SEA antibodies after treatment was also observed in SCS patients (Haribhai et al. 1991; Magalhães-Santos et al. 2003). Intrathecal IgG synthesis was demonstrated in SCS patients by both estimation of the IgG index (Ferrari et al. 1999) and identification of oligoclonal bands in the CSF that were absent in paired serum samples (Pammenter et al. 1991). Higher concentrations of IgG1 in the CSF of SCS patients when compared to the paired serum levels was also observed (Magalhães-Santos et al. 2003). An index based on the ratio between CSF and serum levels of antibodies against schistosome antigens was developed with the aim of distinguishing SCS cases from other CNS disorders (de Jongste et al. 2010), but this index has been evaluated in only one patient affected by SCS.
Taken together, these findings provide strong evidence that methods involving the search for anti-schistosome antibodies in the CSF are likely to be useful for the diagnosis of SCS, and even the encephalic forms. However, they need further investigation. It is necessary to standardize and validate the immunoassays in other populations of NS patients to assess their actual diagnostic value, as well the most effective test or combination of tests. Techniques to detect parasite antigens in the CSF should also be considered. As NS is an uncommon condition, multicentric studies could facilitate the achievement of the adequate sample size.
Currently, a definite diagnosis of SCS can only be established by the demonstration of the eggs and granulomas in nervous tissue. As spinal cord biopsy should be avoided because of the risks (Case 1996; Ferrari 1999), the diagnosis of this disorder is essentially presumptive and based on evidence of spinal cord (low thoracic, lumbar and/or sacral) and/or cauda equina lesions, confirmation of active schistosome infection by a direct method, and exclusion of other causes of myeloradiculopathy (Center for Disease Control and Prevention 1984; Ferrari et al. 1993). This reinforces the need of a non-invasive test (or tests) to enable a more accurate diagnosis of this disease.
5 Treatment and Outcome
Treatment of NS is based on the administration of an antischistosomal drug—usually praziquantel (PZQ)—associated with a corticosteroid. PZQ kills adult worms preventing de novo egg deposition and subsequent granuloma formation. The steroid counters the inflammatory response, reducing compression and destruction of the nervous tissue. No consensus has been reached to date on the optimal therapeutic regimen to treat the different forms of NS. Controlled trials to access the efficacy of different therapeutic regimens have not been hitherto performed. However, some general recommendations can be made.
ASE should be treated initially with steroid (e.g., prednisone 1 mg/kg/day) to suppress the hypersensitivity reaction, followed by PZQ. The optimal timing for this drug is still unclear. Based on rare reports of clinical worsening of patients with ASE after taking PZQ (Jauréguiberry et al. 2007), it has been suggested to prescribe this drug soon after the stabilization of the neurological picture, when the patient is still receiving high steroid doses to avoid clinical worsening (Ferrari and Moreira 2011). As immature worms are not susceptible to PZQ, a second run of administration should be given after 6–12 weeks (Gryseels et al. 2006).
Although either surgical resection (followed by the use of PZQ) or medical therapy alone has been successfully used to treat PES, the clinical treatment is preferred (Fowler et al. 1999). PZQ and a steroid (equivalent to 1 mg/kg/day of prednisone) should be promptly started, and the steroid should be continued upon clinical monitoring.
The common SCS treatment schedule is Brazil foresees PZQ (60 mg/kg/day for 3 days—maximum daily and total dose of 5 g and 15 g, respectively) administered in two daily doses at a 4-h interval, and intravenous methylprednisolone (15 mg/kg/day for 5 days—maximum dose 1 g/day) divided into two daily doses followed by prednisone (1.5–2 mg/kg/day) given in three daily doses for about 3–4 weeks, and then replaced with a single daily dose, which is gradually tapered until its complete discontinuation within 3–4 months (Ferrari 2004; Ferrari et al. 2008a). The steroid should be started immediately in highly suspected cases and PZQ soon after the confirmation of schistosome infection.
Surgical treatment of SCS should be limited to highly selected cases particularly those with evidence of spinal compression and rapidly worsening of motor function in lower limbs despite medical treatment (Ferrari and Moreira 2011; Carod-Artal 2012).
Related posts:
 Health Problems Associated with the Use and Abuse of Khat (Catha edulis)
Health Problems Associated with the Use and Abuse of Khat (Catha edulis)
 African Trypanosomiasis: A Highly Neglected Neurological Disease
African Trypanosomiasis: A Highly Neglected Neurological Disease
 Neurology of a Permanent and Non-progressive Motor Neuron Disorder Associated with Food (Cassava) Toxicity
Neurology of a Permanent and Non-progressive Motor Neuron Disorder Associated with Food (Cassava) Toxicity
 Preservation, Snake Venoms and Stroke in the Tropics
Preservation, Snake Venoms and Stroke in the Tropics
 of Khat (Catha edulis Forsk)
of Khat (Catha edulis Forsk)
 Infections: Neurological Involvement and Neurobiology
Infections: Neurological Involvement and Neurobiology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


