5 Neurosurgery A. Aneurysms 1. Epidemiology a. Adult prevalence 2% b. Annual incidence of aneurysmal subarachnoid hemorrhage (SAH) ~6–8/100,000, peak age 50s c. Modifiable risk factors for SAH—hypertension, smoking, excessive alcohol 2. Presentation/Natural history a. Saccular aneurysms (1) 10–15% of SAH fatal before reaching hospital, ~50% by 1 month (2) Overall, ~⅓ die, ⅓ have severe permanent disability, and ⅓ return to baseline. (3) Major morbidities include rebleeding, hydrocephalus (~15–20%), cardiac (up to 50%), vaso-spasm, hyponatremia, and seizures. (4) See Tables 5.1 – 5.4 for risk of rupture data and grading scales. b. Fusiform aneurysms (1) Treatment options are medical (antiplatelet therapy or anticoagulation for emboli) and surgical (trapping with or without bypass or resection with vessel reconstruction).
Cranial Procedures
I. Vascular Disease

Grade | GCS | Motor Deficit |
|---|---|---|
I | 15 | No |
II | 13–14 | No |
III | 13–14 | Yes |
IV | 7–12 | Yes or no |
V | 3–6 | Yes or no |
Abbreviations: GCS, Glasgow coma score.
Grade | CT Findings | Incidence of Clinical Vasospasm (%) |
|---|---|---|
I | No hemorrhage evident | 0 |
II | Diffuse SAH with vertical layers < 1 mm thick | 0 |
III | Localized clots and/or vertical layers of SAH > 1 mm thick | 95 |
IV | Diffuse or no SAH, but with intracerebral or intraventricular hemorrhage | 0 |
Abbreviation: SAH, subarachnoid hemorrhage.
c. Dissecting aneurysms
(1) Antiplatelet or anticoagulants if extracranial and associated with ischemia, surgical or endovascular obliteration if intracranial and associated with hemorrhage
d. Traumatic aneurysms
(1) Pseudoaneurysms from arterial rupture and containment of hematoma by surrounding tissue, associated with penetrating (more common) or blunt head injury, can lead to carotid cavernous fistulas (CCFs)
(2) Often present with delayed hemorrhage (but variable), treated surgically or endovascularly
e. Infectious (mycotic) aneurysms
(1) Rare, usually distal, occur in 5–15% with bacterial endocarditis
(2) Most fusiform and friable, may cause stroke or hemorrhage
(3) Usually resolve within 6 weeks with antibiotics. If associated with hemorrhage, is enlarging, or not resolved on follow-up angiogram after antibiotic treatment, treatment is needed.
3. Treatment
a. Endovascular coiling or neurosurgical clipping—Goal is exclusion of aneurysm from circulation; second line options include trapping or parent vessel sacrifice.
b. Randomized controlled trial (RCT) of surgery versus coiling for ruptured intracranial aneurysms (International Subarachnoid Aneurysm Trial [ISAT]) found 31% surgical and 24% endovascular patients dead or dependent at 1 year so coiling is a good option for some cases, but durability of coiling and generalizability of results remain uncertain.
c. For unruptured aneurysms, natural history must be weighed against mortality (1–2% for surgery/coiling) and morbidity (4% coiling and 8% surgery, but these are variable).
d. Most consider endovascular for posterior circulation, surgery for middle cerebral artery (MCA), variable for anterior cerebral artery (ACA) and other sites.
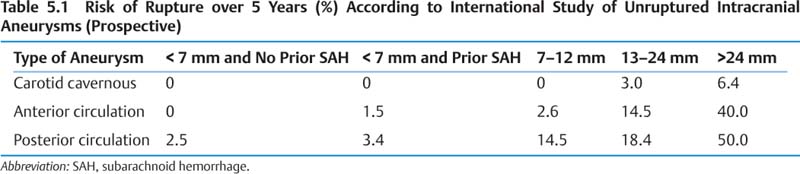
e. Surgical approaches are pterional for most anterior circulation aneurysms; anterior interhemispheric can be used for anterior communicating artery and must be for more distal ACA aneurysms (Fig. 5.1). Posterior circulation aneurysms at basilar apex can be approached by pterional, subtemporal, or combined approach.
B. Arteriovenous malformations (AVMs)
1. Epidemiology
a. Prevalence 15–18/100,000, incidence 1/100,000, typically present before age 40
b. Account for 1–2% of all strokes (3–4% in young adults)
2. Presentation/Natural history
a. Presentations include hemorrhage (50%), seizures (25%), headache, and incidental, focal deficits (due to arterial steal or venous hypertension)
b. Hemorrhage rate 2–4% per year, rehemorrhage rate may be higher. Future hemorrhage is associated with hemorrhagic presentation, large size, deep venous drainage, associated aneurysms.
c. Hemorrhage mortality 10–30%, morbidity 10–30% d. See Tables 5.5 and 5.6 for Spetzler–Martin grading system and associated outcomes.
Fig. 5.1 (A) Right posterior communicating artery aneurysm exposed through a pterional approach. (A) optic nerve, (B) posterior communicating artery, (C) superior hypophy-seal artery, (D) anterior cerebral artery, (E) anterior choroidal artery, (F) middle cerebral artery. (B) Same view after clipping of the aneurysm. (From Alleyne Jr. CH. Neurosurgery Board Review. New York, NY: Thieme; 1997. Reprinted by permission.)
Feature | Points |
|---|---|
Nidus size (cm) | |
Small (< 3) | 1 |
Medium (3–6) | 2 |
Large (> 6) | 3 |
Eloquence of adjacent brain | |
Noneloquent | 0 |
Eloquent (sensorimotor, language, visual, thalamus, hypothalamus, internal capsule, brainstem, cerebellar peduncles, deep cerebellar nuclei) | 1 |
Pattern of venous drainage | |
Superficial only | 0 |
Deep | 1 |
a. Medical management (antiepileptics, etc.), embolization (cure rate generally low, but > 80% if pure fistula or single feeding artery), radiosurgery (hemorrhage risk still present during the ~2–3 years until obliteration, generally considered for < 3 cm nidus with deep and/or eloquent location), surgical excision, or combination of modalities
C. Dural arteriovenous fistulas (DAVFs)
1. Epidemiology
a. 10–15% of intracranial vascular malformations, incidence 0.2/100,000, slight female preponderance, typically present in midlife
2. Presentation/Natural history
a. Clinical presentations include incidental, tinnitus, headache, visual impairment, hemorrhage. Borden I generally benign (conversion rate to higher grade ~2%), but occasionally symptoms warrant treatment. Borden II and III have 15% annual event rate (8% hemorrhage) with 10% annual mortality rate and generally require treatment. See Table 5.7 for classification of DAVF III:
3. Treatment
a. Goal is to eliminate cortical venous reflux (CVR). Treatment options include endovascular (trans-arterial embolization – rarely cures, versus transvenous coiling – generally preferred), surgical disconnection of CVR, and radiosurgery (~⅔ cure rate, generally third choice modality).
D. Moyamoya disease
1. Epidemiology
a. Incidence—rare (< 1/100,000), though higher in Japan
b. Two peaks at mean ages 3 and 20s–30s
c. Often linked to inflammatory conditions, also associated with atherosclerosis
2. Presentation/Natural history
a. Typical presentations are ischemic (80% in pediatric cases), hemorrhage (60% in adult cases), seizures, progressive cognitive decline. 73% major deficit or death within 2 years of diagnosis in children. Treatments address ischemia with less effect on hemorrhage risk; good prognosis in ~60%.
3. Treatment
a. Surgical revascularization including direct bypass, indirect revascularization (encephalomyosynangiosis, encephaloduromyosynangiosis), omental transposition, multiple burr holes
1. Epidemiology
a. Prevalence 0.5%, incidence 0.5/100,000/year, three autosomal dominant familial forms known (CCM1–3, particularly prevalent in Hispanic population)
2. Presentation/Natural history
a. Presenting symptom seizures (50%, incidence 1–2%/year), focal deficit (25%, ~ 40% fully resolve), incidental, headaches. Risk of symptomatic hemorrhage 0.5–2%/year, may be higher in patients with previous hemorrhages (~ 5%/year), deep lesions (~10%/year), posterior fossa lesions, familial inheritance, and women (~ 4%/year).
3. Treatment
a. Observation/medical for control of seizures, surgical excision has good–excellent outcome in ~90%, lower with deep lesions; radiosurgery does not alter radiographic appearance and has not been shown to alter the natural history
F. Intracerebral hemorrhage (ICH)—no structural lesion
1. Epidemiology
a. Incidence 12–15/100,000/year, 15–30% of all strokes
b. Risk factors include age > 60, male sex, heavy ethanol use, drugs (cocaine, etc.), coagulopathy, hypertension (most common cause), amyloid angiopathy (most common cause in elderly)
2. Presentation/Natural history
a. Poor prognosis with age > 60, increasing hematoma volume (particularly > 30 cc, Table 5.8), lower initial Glasgow Coma Scale (GCS), basal ganglia or brainstem (deep) location, presence of intraventricular hemorrhage (IVH)
Volume (0.5 ABC) (cc) | 30-Day Mortality (%) | Functional Independence (%) |
|---|---|---|
< 30 | 23 | 18.0 |
30–60 | 64 | 1.4 (all cases > 30 cc) |
> 60 | 93 |
Abbreviation: 0.5ABC = method for measuring volume of hematoma as half the length times width times height on CT scan.
3. Treatment
a. Medical management (target systolic blood pressure < 180 mm Hg, correct coagulation, control increased intracranial pressure [ICP]) or surgical evacuation. Surgery for supratentorial ICH not proven to benefit, decreases mortality without changing morbidity in putamen/thalamic hemorrhages, and evacuation of superficial hemorrhages may be life saving but may not alter recovery of deficits. Surgery may benefit patients < age 60 with lobar ICH, < 1 cm from surface and with initial GCS > 8. Cerebellar hemorrhage > 3 cm, neurologically deteriorating, brainstem compression, or obstructive hydrocephalus should have urgent evacuation.
G. Infarction (see Chapter 3 section XXI and Chapter 4 section XIX)
1. Natural history for surgical stroke
a. Cerebellar infarct—Postinfarct edema can lead to brainstem compression and obstructive hydro cephalus (risk mainly 12–96 hours postictus, mortality up to 80%).
b. MCA territory infarct—“malignant” cerebral edema in up to 10%, mortality up to 80%
2. Treatment
a. Cerebellar infarct—Early symptoms warrant emergent craniectomy/removal infarcted tissue, can be life saving with good outcome if brainstem not involved.
b. MCA territory infarct—Early hemicraniectomy can reduce mortality to ~30–40%, but morbidity generally severe. Better outcomes (option) in young patients with nondominant hemispheric involvement.
H. Carotid stenosis
1. Epidemiology
a. Prevalence (any degree): 2.5% < age 65, 35% > age 75
b. Prevalence (> 50% stenosis): < 5% in general adult population, 10% of age > 60 with ≥ 1 cardiovascular (CV) risk factor (↑ with ↑ age and number of CV risk factors)
2. Presentation/Natural history (Table 5.9)
a. Presentations include transient ischemic attack (TIA), stroke, and amaurosis fugax.
b. Recurrent or post TIA/stroke risk is front loaded: 20% are in first month.

3. Treatment—medical management (Chapter 4 section XIX)
a. Carotid endarterectomy generally appropriate for symptomatic patients with 70–99% stenosis, a consideration for symptomatic patients with 50–69% stenosis or asymptomatic patients with 60–99% stenosis (careful patient selection and low perioperative morbidity and mortality). Timing of surgery is preferable within a few weeks (↓ upfront stroke risk).
b. Endovascular angioplasty ± stenting is under investigation; generally considered an option in high-risk patients where early benefit appears similar to surgery.
I. Arterial dissections
1. Epidemiology
a. Associated with fibromuscular dysplasia, connective tissue diseases
b. Intracranial
(1) Traumatic/iatrogenic: less common than spontaneous
(2) Spontaneous— ~70% vertebrobasilar, account for < 10% spontaneous SAH (mainly age > 30), rare cause of stroke (mainly age < 30)
(1) Traumatic/iatrogenic: more common than spontaneous
(2) Spontaneous—more common in young women
2. Presentation/Natural history
a. Typical presentations include headache/neck pain, Horner syndrome (for internal carotid artery [ICA]), TIA/stroke
b. Traumatic generally present < 24 hours
c. Intracranial vertebrobasilar dissections—if presenting with hemorrhage, rebleed rate up to 30–70% with 70% occurring in first 24 hours, 80% in first week, 90% in first month; can also cause symptomatic embolization/stenosis; overall mortality ~30%.
3. Treatment
a. Extracranial—anticoagulate for 6–12 weeks, antiplatelet agents, or if emboli persist, repair of vessel with interposition vein graft, bypass, or endovascular angioplasty with or without stenting
b. Intracranial—similar except if SAH presentation; requires preventative treatment for future hemorrhage. Surgical options include proximal clipping if collateral flow adequate, trapping, resection with or without bypass. Endovascular stenting or occlusion of dissection is also a viable option.
J. Carotid cavernous fistula (CCF)
1. Epidemiology
a. Traumatic—prevalence in head trauma 0.2%
b. Spontaneous—includes direct high-flow (typically ruptured cavernous ICA aneurysms), low-flow dural shunts from meningeal branches of ICA, external carotid artery (ECA), or both
2. Presentation/Natural history
a. Orbital/retroorbital pain, chemosis, pulsatile proptosis, ocular/cranial bruit, visual deterioration, diplopia, and ophthalmoplegia
b. 50% of low-flow CCFs spontaneously thrombose.
3. Treatment
a. Low-flow CCFs can be watched until they spontaneously thrombose if visual acuity stable and intraocular pressure < 25 mm Hg
(1) Endovascular embolization transarterially through ICA or ECA (placing balloon in fistula or trapping with two balloons ± bypass) or transvenous coiling via petrosal sinus (from the jugular vein) or superior ophthalmic vein (enter supraoptic vein as it enters orbit to become superior ophthalmic vein, Fig. 5.2).
(2) Rarely open surgery
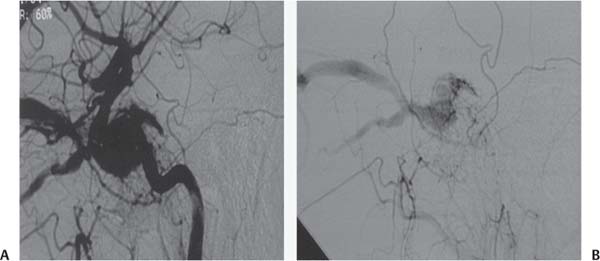
Fig. 5.2 Carotid cavernous fistula (CCF) seen on lateral angiography of internal carotid artery injection. (A) Early filling of the cavernous sinus in the arterial phase with dilated superior and inferior ophthalmic veins and (B) filling from the external carotid artery.
II. Infectious Disease
A. Cerebral abscess
1. Epidemiology
a. Prevalence— ~2500 cases/year in U.S.; male:female (M:F) ratio is 2:1
b. Contiguous spread (most common, 40%)—sinusitis, otitis media, dental abscess
c. Hematogenous spread—lung infections, pulmonary AVF, congenital cyanotic heart disease, bacterial endocarditis, immunodeficiency, dental abscess, gastrointestinal infections
d. Penetrating source—penetrating head trauma, neurosurgery, cerebrospinal fluid (CSF) leak
e. Pathogens include Streptococcus (most common), Streptococcus milleri and anginosis (sinusitis), Bacteroides, Proteus, Staphylococcus aureus (trauma), Staphylococcus epidermidis (iatrogenic), Actinomyces (dental), fungal (immunocompromised)
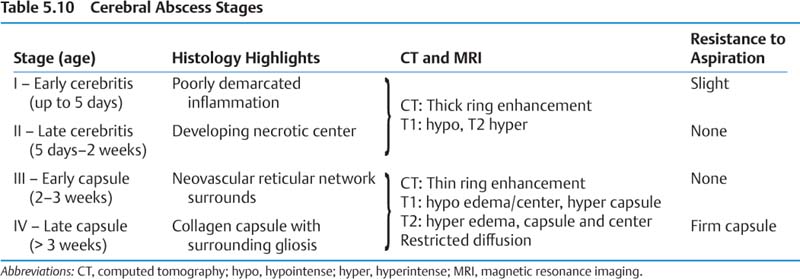
2. Presentation/Natural history (Table 5.10)
a. Symptoms nonspecific for brain lesion, history typically acute
b. Mortality 10–20%, neurologic disability 45%
3. Treatment
a. Antibiotics—can be sole treatment for cerebritis stage (< 2 weeks of symptoms), < 3 cm diameter, known organism; typically at least 6 weeks intravenous (IV) therapy
b. Surgical indications—significant mass effect/elevated ICP, no improvement after 4 weeks of antibiotics, proximity to ventricle (increased likelihood of ventricular rupture), uncertain diagnosis of pathogen, trapped foreign material, and poor condition of patient
c. Surgical options—stereotactic or computed tomographically (CT) guided aspiration; craniotomy and excision if encapsulated or foreign material
B. Subdural empyema/Epidural abscess
1. Epidemiology
a. 5 times less common than cerebral abscess, subdural empyema more common than epidural abscess, M:F ratio 3:1
b. Generally from contiguous spread or penetrating source (rarely hematogenous), can be secondary to meningitis
c. Associated cerebral abscess in 25% cases
2. Presentation/Natural history
a. Presents as nonspecific brain lesion, meningismus common, more commonly febrile than with cerebral abscess alone
b. Subdural empyema often leads to cortical venous infarct, secondary cerebritis/abscess
c. Mortality 10–20%, morbidity ~50%, worse prognosis if age > 60 and poor presentation
3. Treatment
a. Rarely antibiotic therapy alone (if patient well, collection is small, early response), multiple burr holes for drainage (if early), or craniotomy/debridement
C. Shunt infection
1. Epidemiology
a. ~5% risk of early infection
b. < 5% risk of late infection (> 6 months), typically seeding from septicemia
c. Risk factors—length of procedure, younger age
d. Pathogens—Staphylococcus epidermidis (60–75%), Staphylococcus aureus, gram-negatives
2. Presentation/Natural history
a. Presents as systemic infection, abdominal pain, obstruction, tenderness along tubing
b. Mortality 10–15%. Shunt nephritis (immune complex deposition in renal glomeruli), usually with ventriculoatrial shunts, rarely occurs.
3. Treatment
a. Antibiotics alone—poor success, considered only in high surgical risk, terminally ill patients. Slit ventricles are difficult to catheterize in a highly shunt-dependent patient.
b. Typically requires complete hardware removal, external drainage, IV antibiotics (typically vancomycin first line if gram-positive); await ≥ 3 days of sterile CSF, then place new shunt
D. Osteomyelitis/Infected bone flap
1. Epidemiology
a. Very rare in absence of surgery (producing initially avascular bone flap), can result from contiguous spread or penetrating trauma (hematogenous rare)
b. Staphylococcus aureus most common, followed by Staphylococcus epidermidis
2. Presentation/Natural history
a. Variable, progressive/erosive
b. Gradenigo syndrome (Chapter 3 section X)
3. Treatment
a. Antibiotics alone rarely curative, generally requires surgical debridement/removal bone flap, 6–24 weeks antibiotics, cranioplasty at > 6 months
III. Neoplastic Disease
A. Approaches
1. Inferior frontal lobe and parasellar region—bicoronal incision with unilateral or bilateral subfrontal approach or pterional approach
3. Frontal lobe—linear, curved, or horseshoe incision with frontal craniotomy
4. Anterior temporal lobe—linear incision with temporal craniotomy
5. Posterior temporal lobe—linear, reverse question mark, or Isle of Mann incision with temporal craniotomy
6. Parietal lobe—linear or horseshoe incision with parietal craniotomy
7. Occipital lobe—linear or horseshoe incision with occipital craniotomy
8. Trigone of the lateral ventricle—linear or horseshoe incision with appropriate craniotomy for superior parietal, middle temporal gyrus, lateral temporooccipital, or transoccipital approach
9. Anterior third ventricle—linear or horseshoe incision with frontal parasagittal craniotomy and interhemispheric/transcallosal or transcortical approach. The third ventricle can then be approached through the interforniceal or transchoroidal (displace the choroid plexus laterally, divide the tela choroidea, and enter the foramen of Monro between the choroid and fornix) approaches (Fig. 5.3).
10. Posterior third ventricle/pineal region—linear or horseshoe incision with (1) suboccipital transtentorial approach, (2) supracerebellar infratentorial approach, (3) interhemispheric transcallosal (splenium) approach, and (4) transcortical parietal approach (seldom used).
11. Midline posterior fossa/fourth ventricle—linear incision with sub-occipital craniotomy
12. Lateral posterior fossa/CPA—linear incision with retrosigmoid craniotomy
13. Upper clivus—linear or horseshoe incision with subtemporal approach and anterior petrosectomy
14. Middle and lower clivus—curvilinear incision with combined retrosigmoid posterior temporal craniotomy and posterior petrosectomy
B. Localization
1. Nasion—located at the midline frontonasal suture
2. Glabella—the most forward point on the midline supraorbital ridge
3. Pterion—located at the junction of the frontal, parietal, temporal, and greater wing of sphenoid bones. It is located two fingerbreadths above the zygomatic arch and a thumb’s breadth behind the frontal process of the zygomatic bone.
4. Asterion—located at the junction of the lambdoid, occipitomastoid, and parietomastoid sutures. It lies on top of the lower half of the transverse/sigmoid sinus junction.
5. Lambda—located at the junction of the lambdoid and sagittal sutures
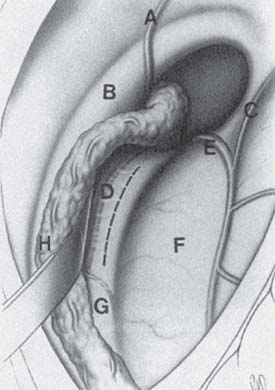
Fig. 5.3 Anatomy of the right foramen of Monro. (A) Septal vein, (B) column of fornix, (C) anterior caudate vein, (D) tela choroidea, (E) thalamostriate vein, (F) thalamus, (G) internal cerebral vein, (H) choroid plexus. (From Alleyne Jr. CH. Neurosurgery Board Review. New York, NY: Thieme; 1997. Reprinted by permission.)
6. Bregma—located at the junction of coronal and sagittal sutures
7. Inion—located at the indentation under the external occipital protuberance that overlies the torcula
8. Opisthion—located at the posterior margin of the foramen magnum in the midline
9. Sylvian fissure—located by (1) marking the 75% point on a line over the superior sagittal sinus from the nasion to the inion; (2) marking the frontozygomatic point, which is 2.5 cm up along the orbital rim past the zygomatic portion; and (3) the sylvian fissure extends along the line connecting the 75% point and the frontozygomatic point. The pterion is located 3 cm behind the frontozygomatic point along the sylvian line.
10. Rolandic fissure—located by (1) the upper rolandic point, with is 2 cm posterior to the 50% point of the midline nasion/inion line; and (2) the lower rolandic point is at the junction of the line from the upper rolandic point to the midzygomatic arch and the sylvian fissure line as previously defined. The rolandic fissure lies between these two points. The lower rolandic point is also 2.5 cm behind the pterion along the sylvian line. The motor strip is usually 4 to 5.4 cm behind the coronal suture.
11. Angular gyrus (part of Wernicke area)—usually just above the pinna, although it is quite variable
C. Neuroepithelial tumors
1. Astrocytic tumors
a. Diffusely infiltrating astrocytomas
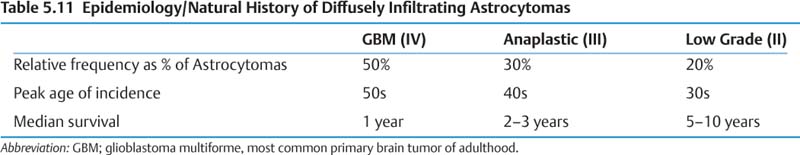
(1) Epidemiology and natural history (Table 5.11)
(a) Dedifferentiation from low grade to high grade occurs sooner with age > 45.
(b) 1–20% present as multiple gliomas.
(c) Gliomatosis cerebri—rare, involving ≥ 2 lobes, poor prognosis
(2) Treatment
(a) Diagnostic surgical biopsy or partial resection recommended in almost all cases. Gross total resection is the best option if tumor location and patient condition permit.
(b) Treatment options for low-grade lesions include serial follow-up, radiation and/or chemotherapy, surgery (no clearly superior strategy). Aggressive treatment may be appropriate for more aggressive tumors (young patients, large tumors that enhance, or patients with short clinical history or progression on imaging).
(c) Standard treatment for high-grade lesions is gross total resection followed by external beam radiation and temozolomide chemotherapy. Poor prognosis with older patients (> 60), glioblastoma multiforme (GBM) histology, poor preoperative (preop) performance status
b. Pilocytic astrocytoma
(1) Epidemiology and natural history (Table 5.12)
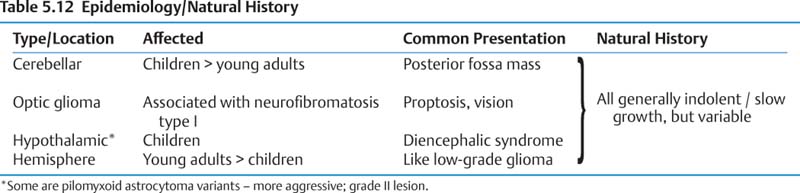
(a) All can cause obstructive hydrocephalus.
(2) Treatment
(a) Cerebellar/hemispheric—treatment of choice is surgical excision. Cyst wall need not be completely removed if nodule is resected.
(b) Hypothalamic/optic—surgical excision if involving single nerve and sparing chiasm, otherwise biopsy + chemotherapy/radiation
2. Oligodendroglioma
a. Epidemiology
(1) ~ 25% of glial tumors, M:F ratio 3:2, mean age 40
b. Presentation/Natural history
(1) Often present with seizures and/or hemorrhage, nonspecific mass effect
(2) 5-year survival rate 40–70% (grade dependent), overall median 3 years postoperation
(3) Oligoastrocytoma variant behaves similarly to oligodendroglioma; both have more aggressive anaplastic forms.
c. Treatment
(1) Surgical resection followed by chemotherapy (favorable response rate associated with allelic losses of chromosomes 1p and 19q)
(2) Radiation for anaplastic transformation
3. Ependymoma
a. Epidemiology
(1) 5% of intracranial gliomas, 70% pediatric (peak age is 10–15 years), commonly fourth ventricle floor
(2) Subependymoma is rare form, generally incidental in older patients, rarely surgical
b. Presentation/Natural history
(1) Generally presents as slow-growing posterior fossa mass, anaplastic form more aggressive
(2) Up to 80% 5-year survival in treated young patients, 40% in those < age 4 or elderly
c. Treatment
(1) Maximal possible resection (extent affects survival) followed by fractionated radiation
(2) Spinal magnetic resonance imaging (MRI) + lumbar puncture for cytology to rule out subarachnoid metastases; spinal radiation if positive
4. Choroid plexus papilloma and carcinoma
a. Epidemiology
(1) 1% of intracranial tumors, 70% occur at age < 2. Majority are benign papillomas.
b. Presentation/Natural history
(1) Typically present with hydrocephalus
c. Treatment
(1) Benign lesions require total surgical excision and adjuvant chemotherapy; use radiation only for carcinoma.
5. Pediatric brainstem gliomas
a. Epidemiology
(1) 10–20% pediatric brain tumors, mean age 7
b. Presentation/Natural history
(1) Tectal glioma—typically present with hydrocephalus, otherwise very benign, 80% 5-year progression-free survival rate
(2) Focal tegmental mesencephalic—often present with hemiparesis, slowly progress
(3) Diffuse pontine glioma—present with multiple cranial nerve palsies, ataxia, increased ICP, median survival < 1 year
c. Treatment
(1) Tectal gliomas require vigilant follow-up and often CSF diversion. Focal tegmental mesencephalic tumors are resected with adjuvant chemotherapy and radiation if they recur. Radiation +/− experimental chemotherapy and/or palliative are used for diffuse pontine glioma.
6. Other low-grade glial tumors
a. Angiocentric glioma
(1) Rare, slow growth, typically present with seizures in children/young adults. Resection is generally curative.
b. Chordoid glioma of the third ventricle
(1) Rare, typically in adults causing hydrocephalus/chiasm compression/hypothalamic dysfunction, resection generally curative
c. Astroblastoma
(1) Rare, mainly in children/young adults, surgically resected, adjuvant radiation/chemotherapy if rare high-grade form
7. Neuronal and mixed neuronal-glial tumors
a. Lhermitte–Duclos disease (dysplastic cerebellar gangliocytoma)
(1) Epidemiology
(a) Rare, affects young adults (mean age 34), associated with Cowden syndrome (multiple hamartomas)
(2) Presentation/Natural history
(a) Typically present with increased ICP/hydrocephalus, cerebellar signs, occasionally mental retardation
(b) May progress slowly
(3) Treatment
(a) Surgical resection
b. Desmoplastic infantile ganglioglioma (DIG)
(1) Epidemiology
(a) Virtually all < age 2 (peak 3–6 months), M:F ratio 2:1
(2) Presentation/Natural history
(a) Commonly present with increased head size, bulging fontanelles, paresis, seizures
(b) Median survival > 75% at 15 years, anaplasia very rare
(a) Complete surgical resection curative for most, chemotherapy for anaplasia
c. Dysembryoplastic neuroepithelial tumor (DNET)
(1) Epidemiology
(a) < 1% primary brain tumors, affects children and young adults (< age 20)
(2) Presentation/Natural history
(a) Typically presents with epilepsy, benign with no or very slow growth
(3) Treatment
(a) Surgical resection of lesion ± neighboring epileptogenic foci
d. Ganglioglioma/gangliocytoma
(1) Epidemiology
(a) < 12% intracranial tumors, generally present < age 30 (peak 11)
(2) Presentation/Natural history
(a) Typically presents with epilepsy, benign, slow growth
(b) 5–10 year survival, 80–90% with treatment
(3) Treatment
(a) Complete surgical excision, radiation considered for rare anaplastic ganglioglioma
e. Central neurocytoma
(1) Epidemiology
(a) Rare, ~10% intraventricular neoplasms (rarely extraventricular), 75% in ages 20–40
(2) Presentation/Natural history
(a) Typically present with increased ICP/hydrocephalus, seizures
(b) Benign, slow growing, rarely hemorrhage, 5-year survival rate > 80%
(3) Treatment
(a) Resection usually cures, stereotactic radiosurgery/chemotherapy options if rare recurrence
f. Cerebellar liponeurocytoma
(1) Epidemiology
(a) Rare adult tumor of posterior fossa
(2) Presentation/Natural history
(a) Presents as posterior fossa mass, behaves as World Health Organization (WHO) grade II lesion
(3) Treatment
(a) Complete surgical excision, radiation may prevent recurrence
g. Papillary glioneuronal tumor
(1) Epidemiology
(a) Rare adult tumor
(2) Presentation/Natural history
(a) Typically presents with seizures, benign with slow growth
(3) Treatment
(a) Complete surgical excision generally curative
h. Rosette-forming glioneuronal tumor of fourth ventricle
(1) Epidemiology
(a) Rare tumor in young adults
(2) Presentation/Natural history
(a) Typically presents with hydrocephalus and ataxia, indolent behavior, recurrence rare
(3) Treatment
(a) Surgical excision generally curative
(1) Epidemiology
(a) Rare, Table 5.13 lists specific names associated with location.
Site | Designation |
|---|---|
Carotid bifurcation | Carotid body tumor |
Superior vagal ganglion | Glomus jugulare tumor |
Auricular branch of vagus | Glomus tympanicum |
Inferior vagal ganglion | Glomus intravagale |
Adrenal medulla and sympathetic chain | Pheochromocytoma |
(2) Presentation/Natural history
(a) Presents as slow growing mass, systemic features of catecholamine release, carcinoid-like syndrome with cranial nerve palsies related to location
(b) Slow growth, benign, 5-year survival rate ~90%, rarely hemorrhage
(3) Treatment
(a) Medical therapy includes α/beta blockers to prevent blood pressure lability and arrhythmias.
(b) Radiation therapy is used generally if surgery is not possible.
(c) Embolization prior to surgery can reduce intraop blood loss. (d) Surgical resection is preferred.
8. Pineal region tumors
a. Pineocytoma
(1) Epidemiology
(a) < 1% primary brain tumors, mainly children and young adults (peak incidence age 10–20)
(2) Presentation/Natural history
(a) Typically present with increased ICP/hydrocephalus, Parinaud syndrome
(b) Stable or slow growth, 5-year survival rate ~90%, rarely hemorrhage
(3) Treatment
(a) Surgical resection, stereotactic biopsy high risk
b. Pineoblastoma
(1) Epidemiology
(a) < 1% primary brain tumors, along with pineocytomas and intermediate tumors (features of both) account for 15% pineal region tumors, most in children (peak age 3), M:F ratio 1:2
(2) Presentation/Natural history
(a) Typically present with increased ICP/hydrocephalus, Parinaud syndrome
(b) Primitive neuroectodermal tumor (PNET) with CSF seeding in ~ 50%, median survival 2 years
(3) Treatment
(a) Surgical resection + cranial/spinal radiation if age > 3 + chemotherapy
c. Papillary tumor of the pineal region
(1) Epidemiology
(a) Rare tumor in children and young adults
(2) Presentation/Natural history
(a) Typically presents with hydrocephalus and WHO grade II–III behavior; can recur
(a) Surgical resection followed by focal radiation
9. Embryonal/Primitive neuroectodermal tumors (PNETs)
(1) All tend to disseminate via CSF (often into the spinal subarachnoid space), 20–50% at diagnosis
a. Medulloblastoma
(1) Epidemiology
(a) 15–20% of total and ~⅓ of posterior fossa pediatric brain tumors, rare in adults, most diagnosed by age 5, M:F ratio 3:1
(2) Presentation/Natural history
(a) Rapid presentation with increased ICP/hydrocephalus, cerebellar signs
(b) Standard risk – no metastases, no gross residual postresection = 5-year survival approaches 100% if ERBB-2 tumor protein negative, 54% if ERBB-2 positive
(c) High risk – metastases, residual postresection, 5-year survival ~ 20%
(3) Treatment
(a) Surgical excision, adjuvant chemotherapy, craniospinal radiation if age > 3
b. CNS PNET/Supratentorial PNET
(1) Epidemiology
(a) 1% pediatric brain tumors, median age 3, M:F ratio 2:1
(b) Variants include CNS neuroblastoma (including esthesioneuroblastoma), ganglioneuroblastoma, medulloepithelioma, pineoblastoma, and ependymoblastoma.
(2) Presentation/Natural history
(a) Presentation varies with site of origin (e.g., seizures if hemispheric, vision/endocrine if suprasellar, hydrocephalus/Parinaud syndrome if pineal).
(b) 30% 5-year survival; survival associated with complete resection, no metastases, age > 2, heavily calcified lesion
(3) Treatment
(a) Aggressive surgical excision, adjuvant chemotherapy, craniospinal radiation if age > 3
c. Atypical teratoid-rhabdoid tumor
(1) Epidemiology
(a) Almost always age < 3
(2) Presentation/Natural history
(a) Presents as raised ICP, developmental regression, seizure, torticollis
(b) Median survival is 6 months.
(3) Treatment
(a) Gross total resection, radiation usually not option as age < 3, experimental chemotherapy
D. Tumors of cranial and paraspinal nerves
1. Schwannoma
a. Presentation/Natural history
(1) 8% of intracranial tumors
(2) Parenchymal typically present with epilepsy or focal deficit before age 30, vestibular schwannoma with sensorineural hearing loss/tinnitus/dizziness after age 30
(3) Slow growth, < 10% recurrence postresection
(4) Can be complicated by hydrocephalus requiring shunt
b. Treatment
(1) Audiology assessment helps treatment decisions/gives baseline.
(2) Can follow symptoms/radiology/audiology every 6 months for < 3 cm lesions
(4) Surgical resection of lesions < 3 cm adds benefit of tumor removal, generally for all > 3 cm, 80% normal/near-normal cranial nerve (CN) VII and up to 40% hearing preservation overall with small vestibular schwannoma (generally total loss with large lesions)
(5) Surgical approaches include translabyrinthine (sacrifices hearing if still present, but may better preserve CN VII), suboccipital (may best preserve hearing), retromastoid, subtemporal
2. Neurofibroma (including plexiform)
a. Epidemiology
(1) Rarely intracranial, plexiform neurofibromas often in orbit (CN V1), scalp, parotid (CN VII)
(2) Associated with neurofibromatosis (especially plexiform in neurofibromatosis type I [NF1])
b. Presentation/Natural history
(1) Typically present as painless mass, slow growth, benign, 2–12% degenerate into malignant peripheral nerve sheath tumor, recurrence rate high
c. Treatment
(1) Surgical resection (complete usually impossible) usually for symptomatic spinal cord or neural compression
3. Other
a. Perineurioma
(1) Rare lesion, rarely intracranial affecting cranial nerves, plexiform variant described
(2) Presents as benign mass, treated with surgical resection
b. Malignant peripheral nerve sheath tumor
(1) Rarely intracranial affecting cranial nerves, can arise from neurofibromas or radiation exposure, malignant/aggressive, treated with surgical resection ± chemotherapy/radiation
E. Tumors of the meninges
1. Meningiomas
a. Epidemiology
(1) Autopsy prevalence 3% in > age 60, 15–20% of primary intracranial tumors (second to GBM)
(2) Multiple in up to 8%, females twice as commonly affected as males, higher in NF, rare in childhood unless NF1
b. Presentation/Natural history
(1) Incidental presentation in up to 50% cases, typically grow slowly, ~1% malignant
(2) Overall 5-year survival > 90%, 20-year recurrence rate 20–50%
(3) Most important risk factors for recurrence are atypical histology and extent of resection (Table 5.14)
Grade | Extent of Resection | Recurrence Rate* (%) |
|---|---|---|
I | Complete including dural attachment and abnormal bone | 10 |
II | Complete with cauterization of dural attachment | 15 |
III | Complete without dural attachment | 30 |
IV | Incomplete resection | Up to 85 |
V | Biopsy | 100 |
*Length of follow-up varies around 5 years; numbers may increase with longer follow-up.
(1) Surgical resection is treatment of choice
2. Benign mesenchymal tumors and tumor-like lesions
a. Epidemiology
(1) Rare lesions affecting meninges and/or adjacent skull/scalp
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


