Fig. 14.1
Magnetic resonance imaging of a patient with malignant glioma. a Axial T1-weighted image reveals water-containing tissue as hypo-intense. Central hypo-intense area is consistent with necrosis. b T2-weighted MRI from the same patient allows water-containing tissue to be seen as hyper-intense. This signal is consistent with vasogenic edema. (Reproduced with permission fromPope 2005. Copyright American Society of Neuroradiology)
14.1.4 Pathological Features of Malignant Gliomas
Pathological examination of suspected neoplastic tissue is required for definitive diagnosis of malignant gliomas. This may sometimes lead to an underestimation of the degree of malignancy because of the heterogeneous nature of malignant gliomas (Rees et al. 1996) . For example, a biopsy may be obtained from a more differentiated portion of the tumor, leading to an underestimation of disease severity. Nevertheless, identification of nuclear atypia , mitotic figures , endothelial cell proliferation, and necrosis are important for grading the level of malignancy. These features are not unique to malignant gliomas and are shared by other cancers of the body. Importantly, current therapies for malignant gliomas exclusively target these neoplastic features shared with other cancers as further detailed later in this chapter.
The WHO grading system for tumors rates the differentiation level of tumors on a scale of I to IV. WHO Grade I tumors typically have a favorable prognosis and are slow growing, not malignant, and lack nuclear atypia, mitotic figures, endothelial cell proliferation, and necrosis. WHO Grade II tumors are less differentiated and typically show nuclear atypia. WHO Grade III and Grade IV tumors are malignant and aggressive cancers showing signs of both nuclear atypia and mitosis. WHO Grade IV cancers in addition can contain areas of necrosis and microvascular proliferation and carry a poor prognosis. Most malignant gliomas are WHO Grade IV (CBTRUS 2011), reflecting the robustly de-differentiated state of individual glioma cells. And as with other WHO Grade IV cancers of the body, malignant gliomas require early and aggressive treatment to prevent uncontrolled growth and spread.
14.1.4.1 Nuclear Atypia and Mitotic Figures
Nuclear atypia, characteristic of WHO Grade II tumors and above, is abnormal nuclear morphology. The nuclei in these cells are pleomorphic, demonstrating variability in shape and size. Typically the nucleus to cytoplasm ratio is 1:4 or 1:6, but in tumor cells, including malignant gliomas, this ratio can approach 1:1. WHO Grade III cancers and above have larger numbers of mitotic figures, the mitotic spindles distinctive of actively dividing de-differentiated cells. Visualization of greater than 1 dividing cell per 10 high power microscopic fields correlates with a decrease in median survival from 5.5 years to 1 year in patients with malignant gliomas (Fulling and Garcia 1985) .
14.1.4.2 Endothelial Cell Proliferation
Vascular endothelial cell proliferation is a common feature among aggressive cancers and portends a poor prognosis. Median survival decreases from 5.5 years to 3.5 years in patients with malignant gliomas when vascular endothelial cell proliferation is observed histologically (Fig. 14.2b) (Fulling and Garcia 1985) . Typically this aberrant proliferation appears as “glomeruloid tufts” of densely packed actively dividing endothelial cells (Jain et al. 2007) and is associated with WHO Grade IV tumors. Tumor cells classically drive endothelial cell proliferation and neovascularization by release of vascular endothelial growth factor (VEGF), the expression of which is induced by hypoxia (Plate et al. 1992; Shweiki et al. 1992) .
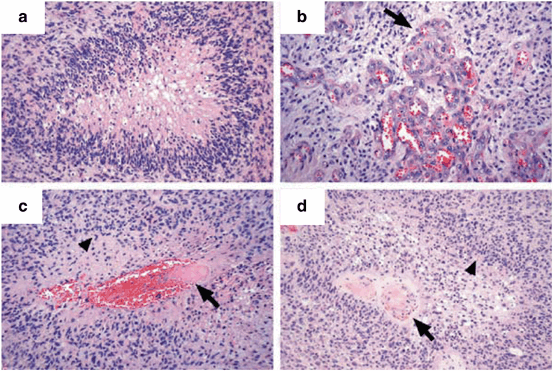
Fig. 14.2
Pathological features of malignant gliomas. a Central area of necrosis surrounded by glioma cells, characteristic of pseudopalisading necrosis. b Arrow points to glomeruloid tufts of densely packed vascular endothelial cells. c, d Arrows points to intravascular thrombosis, likely leading to pseudopalisades (arrowheads) as glioma cells move away from areas of necrosis. (Reproduced with permission from Brat and Van Meir (2004) . Copyright Nature Publishing Group)
Several recent studies have demonstrated that a portion of proliferating endothelial cells present in the tumor mass directly differentiate from glioblastoma stem-like cells. These endothelial cells contain the same mutations present in glioma cells, including amplification of the EGFR gene and chromosome 7 (Wang et al. 2010) . In a separate study, injection of human glioblastoma stem-like cells into immunocomprimised mice lead to tumor vasculature partially composed of human endothelial cells (Ricci-Vitiani et al. 2010) . Additionally, glioma cells were found to trans-differentiate into endothelial cells in a VEGF-independent manner (Soda et al. 2011) . While mouse models demonstrate the importance of glioblastoma stem-like cells in the generation of endothelial cells, a follow-up study of primary human glioblastoma tissue samples showed that more than 90 % of tumor vasculature is derived from endothelial cells and not glioblastoma stem-like cells (Rodriguez et al. 2012) . Thus tumor-derived VEGF may play a more pivotal role in vascular endothelial cell proliferation leading to angiogenesis.
14.1.4.3 Necrosis
High cellular densities, mitotic figures , and nuclear atypia all indicate the robust proliferative ability of glioma cells. In areas of the tumor where cellular density overwhelms the available oxygen tension leading to hypoxia, neovascularization can be triggered secondary to VEGF released by glioma cells. However, these newly formed vessels are disorganized and dilated, hampering adequate perfusion of associated tissues (Fukumura et al. 2010) . As compared to the peritumoral environment, total vascular density, functional vascular density, and total vascular perfusion is significantly reduced in the tumor core; at the center of large tumors, less than 25 % of vessels may effectively perfuse adjacent tissue (Vajkoczy and Menger 2000). Coupled with increased vascular permeability in tumor-associated vessels (Yuan et al. 1994) due to an impaired blood-brain barrier and increased susceptibility to vascular thrombosis (Fig. 14.2c, 14.2d) (Brat et al. 2004), tumor-associated vasculature is hypofunctional (Vajkoczy and Menger 2000) .
Hypofunctional vessels in glioma tumor masses in turn lead to hypoxia-induced necrosis (Brat et al. 2004), a radiological and histological hallmark of Grade IV gliomas. Glioma cells surrounding necrotic tumor cores are up to 20 times more likely to undergo apoptosis and 50 times less likely to proliferate (Brat et al. 2004). These surrounding glioma cells are also more likely to migrate away from the necrotic core, as hypoxia increases the migratory ability of glioma cells (Brat et al. 2004). A central area of hypocellular necrosis surrounded by a rim of glioma cells is known as “pseudopalisading necrosis,” because the migrating hypercellular glioma cells form a “palisade” around a hypoxic necrotic core (Fig. 14.2a). Pseudopalisading necrosis is distinctive of the most aggressive cancers and can be molecularly verified by upregulation of hypoxia inducible factor-1 (HIF-1), which is a transcription factor activated in cells exposed to low oxygen tension. HIF-1 controls the expression of a variety of substrates, including proteins which promote angiogenesis (e.g., VEGF and angiopoietin-1) and glioma cell migration (e.g., C-X-C chemokine receptor type 4 and matrix metalloproteinase-2) (Kaur et al. 2005; Zagzag et al. 2006) . Thus dysfunctional vasculature associated with high-grade gliomas can lead to hypoxia and necrosis, promoting aggressive disease progression.
14.1.5 Molecular Insight Reveals the Heterogeneity of Malignant Gliomas
Recent work by The Cancer Genome Atlas Network has demonstrated that glioblastoma multiforme is a heterogenous disease that can be subdivided into four subgroups based on molecular abnormalities. The (1) Classical subtype is characterized by chromosome 7 amplification and chromosome 10 loss in 100 % of cases examined (Verhaak et al. 2010) . About 97 % of cases also had elevated EGFR amplification, more so than the other three subtypes (Verhaak et al. 2010). There are also a high rate of EGFR mutation and a significantly lower rate of TP53 mutation (Verhaak et al. 2010). Other highly expressed genes in the Classical subtype were NES, NOTCH3, JAG1, and SMO and other genes typical of an astrocytic signature (Verhaak et al. 2010).
Loss of NF1 were characteristic of the (2) Mesenchymal subtype (Verhaak et al. 2010). NF1 mutations frequently occurred together with PTEN mutations (Verhaak et al. 2010). This subtype is so named because of expression of mesenchymal markers, including CHI3L1 and MET (Verhaak et al. 2010). Higher rates of necrosis and inflammatory infiltrates, as well an astrocytic expression signature, characterize this subclass (Verhaak et al. 2010). It is also worth noting that immortalized cell lines most closely matched Mesenchymal tumors (Verhaak et al. 2010).
The (3) Proneural class is characterized by IDH1 point mutations and alterations elevating PDGFR expression (Verhaak et al. 2010). TP53 mutations were also common (Verhaak et al. 2010). Interestingly, genes upregulated in oligodendrocyte development, including OLIG2, PDGFRA, and NKX2–2 were also upregulated in this subclass (Verhaak et al. 2010). Clinically, this subtype was most often found in younger patients and associated with a better prognosis (Verhaak et al. 2010). Finally, the (4) Neural subtype is identified by the presence of neuronal markers, including NEFL, GABRA1, SYT1, and SLC12A5 (Verhaak et al. 2010).
While this subclassification was undertaken to gain a better understanding of genetic mutations that accompany this family of malignancies, the results provide some compelling reasons to speculate that different glioma subtypes arise from different cells of origin and that they may respond quite differently to treatment. For example, these studies have illustrated that the proneural subtype, similar to cells of an oligodendrocyte lineage, does not benefit from current treatment regimens consisting of radiation and chemotherapy, which yield significant decreases in mortality of the classical glioma subtype.
14.1.6 Treatment and Prognosis of Malignant Gliomas
The mainstay of therapy for malignant glioma is aggressive surgical resection, radiation therapy, and temozolomide/Gliadel wafers (Wen and Kesari 2008). Complete surgical resection of malignant glioma cells is nearly impossible given the extraordinary ability of glioma cells to infiltrate unaffected parenchyma and spread throughout the brain. Nevertheless, a retrospective study of 416 consecutive patients with malignant gliomas who were operated on at M. D. Anderson Cancer Center, Houston, Texas between 1993 and 1999 demonstrated that when greater than 98 % of the tumor mass was resected, median survival increased to 13 months as compared to 8.8 months when less than 98 % of the tumor mass was resected (Lacroix et al. 2001) . Thus, aggressive surgical resection is the first clinical step to increasing median survival in patients with malignant gliomas.
Standard radiation therapy is delivered in 30 fractions over a 6-week period for a total dose of 60 Gy (Wen and Kesari 2008) . A randomized study of 303 patients who underwent surgical resection of malignant gliomas demonstrated that radiation therapy can increase median survival from 3.2 months to 8 months (Walker et al. 1978) . More recently, a large randomized study of 573 patients at 85 different centers showed that when radiation therapy is combined with temozolomide, an oral chemotherapeutic agent that alkylates DNA, median survival can be extended from 12.1 months to 14.6 months (Stupp et al. 2005) . Interestingly, the efficacy of temozolomide inversely correlates with the activity of O6-methylguanine-DNA methyltransferase (MGMT), a DNA repair enzyme that removes alkylated DNA. In patients who have epigenetic silencing of MGMT via methylation, median survival increased to 21.7 months upon co-administration of radiation therapy and temozolomide (Hegi et al. 2005) . In lieu of temozolomide, Gliadel wafers can be inserted into the resection cavity to extend median survival from 11.6 to 13.8 months (Westphal et al. 2006) . Gliadel wafers are biodegradable discs containing carmustine, an alkylating agent that can be placed inside the cranium to maximally target unresected tumor cells.
Beyond targeting of the tumor mass, several other comorbidities are treated symptomatically. Seizures, fatigue, cognitive deficits, hyper-coagulability, and depression can all arise in patients suffering from malignant gliomas. Although seizures are treated with anti-epileptic drugs (AEDs), prophylactic administration of AEDs in patients with malignant gliomas is not indicated because of a risk of toxic interactions with chemotherapeutic drugs (Glantz et al. 2000) . Methylphenidate, typically prescribed for attention-deficit hyperactivity disorder, can improve fatigue and cognitive deficits in patients with malignant gliomas (Meyers et al. 1998) . Quite significantly, up to 94 % of patients with malignant gliomas reported symptoms consistent with depression 3 months after surgery, and given that depression correlates with decreased survival and increased medical complications (Litofsky et al. 2004) , a holistic treatment regimen is essential .
14.2 Appreciating the Uniqueness of Gliomas
Although sharing significant histopathological and genetic features with other cancers as detailed in the previous section, malignant gliomas are unique cancers. Unlike other solid cancers of the body, which use intravascular hematogenous routes to metastasize to unaffected organs, malignant gliomas rarely metastasize outside of brain. Instead, glioma cells actively migrate on the abluminal surface of cerebral vessels for tumor expansion. Initially, gliomas are chemotactically drawn to vascular endothelial cells, thereby disrupting the native cytoarchitecture comprising the blood-brain barrier . After vascular engagement, individual gliomas cells migrate away from the tumor core. This dynamic process of cellular migration requires coordinated changes in shape and volume, a process dependent on flux of osmotically-active ions through ion channels and transporters expressed by gliomas.
Also unlike most solid tumors of the body, the growth and expansion of malignant gliomas occurs in an unexpandable space confined by the cranium. Therefore, to create an area of expansion for the growing tumor, glioma cells release glutamate in large concentrations through system xc- , a highly expressed cystine-glutamate exchanger, encoded by theSLC7A11 gene in humans. For every molecule of cystine imported through the channel, one molecule of glutamate is released, raising the concentration of extracellular glutamate to supra-physiological levels. This glutamate acts in an autocrine manner to promote migration of glioma cells, but also induces excitotoxicity in peritumoral neurons, culminating in neuronal death and room for tumor spread. Importantly, this accumulation of large concentration of extracellular glutamate leads to a collection of neurological sequelae strikingly similar to other neurodegenerative diseases . Despite accumulating evidence that malignant gliomas result in similar downstream effects as neurodegenerative diseases, all Food and Drug Administration (FDA)-approved treatment options target features of gliomas shared by other solid cancers of the body. Targeting unique features of gliomas may lead to an improved patient prognosis.
14.2.1 Glioma-Vasculature Interactions
As already mentioned, unlike other solid cancers of the body, malignant gliomas rarely metastasize out of the brain. Less than 2 % of malignant gliomas metastasize outside of the central nervous system , usually occurring late in the disease course (Smith et al. 1969; Beauchesne 2012) . Several hypotheses have attempted to explain why glioma cells poorly spread to extracranial organs. Metastasis typically occurs through three routes: vascular circulation, the lymphatic system, and contact by adjacent structures in a body cavity. To hematogenously spread to other organs, glioma cells must be able to intravasate into the vasculature; however, studies using C6 rat glioma cells demonstrate that glioma cells cannot penetrate the basement membrane associated with cerebral vessels to enter circulation (Bernstein and Woodard 1995) . Alternatively, even if glioma cells entered the vasculature, it is possible that extracranial organs do not contain the milieu of chemoattractants and growth factors necessary to support satellite tumor formation. Also, given that gliomas form in an immuno-privileged environment, movement of glioma cells into systemic circulation may elicit an immune reaction preventing growth outside of the brain (Mourad et al. 2005) . From an anatomic perspective, the dearth of lymphatic vessels in the central nervous system, coupled with dense connective tissue protecting dural veins and a lack of connection between intracranial circulation and the extracranial lymphatic system, prevents the extracranial egress of glioma cells.
Nevertheless, glioma cells are extraordinarily adept at migration and invasion , forming diffuse and expansive intracranial tumor masses. More than 70 years ago, Hans Joachim Scherer, a German neuropathologist, microscopically examined 100 brains of patients with brain tumors and outlined the most common routes of intracranial glioma cell migration and invasion, now known as “Scherer’s structures” (Scherer 1940). Scherer demonstrated that glioma cells in the grey matter congregate around cerebral blood vessels, especially capillaries and other small diameter vessels (Scherer 1940). This collection of glioma cells around vessels is known as “perivascular cuffing” and allows the growing tumor to coopt existing vasculature for its own support. As early as 1 week after implantation, glioma cells injected into rat cerebrum encircled existing vasculature, forming a well-perfused tumor relatively early in the disease course (Holash et al. 1999) . Thus vascular cooption allows the growing tumor to meet its energy demands early in the disease course before the onset of angiogenesis. In white mater, Scherer showed that glioma cells surround myelinated nerve fascicles (Scherer 1940). More recent studies have corroborated Scherer’s pioneering findings and have explored the molecular mechanisms by which the abluminal surface of cerebral vessels and nerve fascicles function as conduits, along which glioma cells migrate away from the tumor core promoting tumor expansion.
14.2.1.1 Glioma Cells Migrate Along Cerebral Vasculature
To migrate along the vasculature, glioma cells must first contact individual vessels. Studies of transplanted C6 rat glioma cells into rat forebrain suggest that migrating glioma cells come into contact with the vasculature in less than 24 h (Farin et al. 2006) . One mechanism by which glioma cells chemotactically migrate towards the vasculature is by responding to bradykinin , a kinin released by vascular endothelial cells (Montana and Sontheimer 2011) . Bradykinin binds to the B2 receptor constitutively expressed by human glioma cells (Montana and Sontheimer 2011). When human glioma cells were placed on murine brain slices, in 2 h bradykinin induced nearly 85 % of glioma cells to contact blood vessels; this interaction was reduced to 30 % when the B2 receptor was selectively knocked down in human glioma cells (Montana and Sontheimer 2011). Bradykinin application also increased the total number of migrating glioma cells, and increased the speed and distance travelled by individual glioma cells (Montana and Sontheimer 2011), further emphasizing the importance of this molecular interaction.
Once contacting the vasculature, glioma cells migrate in a saltatory manner at a speed of about 6–25 µm/h (Farin et al. 2006; Turner and Sontheimer 2013; Watkins and Sontheimer 2011) . Contact is maintained with the vasculature through activation of α-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) glutamate receptors on the leading edges of migrating glioma cells, which promote binding to extracellular matrix components, including type I and type IV collagen found in perivascular areas (Piao et al. 2009) . Increased AMPA receptor expression correlated with enhanced expression of β1-integrins on the plasma membrane and increased numbers of focal adhesion complexes, all of which are involved in attachment of glioma cells to extracellular substrates, promoting glioma cell invasion (Piao et al. 2009).
14.2.1.2 Glioma Cells Disrupt Cytoarchitecture Associated with the Blood-Brain Barrier
The vasculature of the brain is unlike the rest of systemic circulation for at least two principal reasons: (1) tight junctions between vascular endothelial cells form a blood-brain barrier , and (2) the “neurovascular unit,” composed of vascular endothelial cells, astrocytes, and neurons, couples neuronal activity to vascular perfusion. Maintenance of the blood-brain barrier is important, as it prevents ions, molecules, and cells from non-selectively infiltrating the brain, where the extracellular milieu is tightly regulated for optimal neuronal functioning. One mechanism by which neuronal activity leads to enhanced vascular perfusion is through the spillover of glutamate from synapses onto adjacent astrocytes. This glutamate can activate astrocytic metabotropic glutamate receptors (mGluR), which through a cascade involving G-protein coupled signaling lead to downstream increases in intracellular [Ca2+], which in turn leads to release of arachidonic acid metabolites and K+, subsequently modulating of vascular tone (Drake and Iadecola 2007; Attwell et al. 2010) . An increase in cerebral blood flow in response to increased neuronal activity is termed “functional hyperemia” and serves to enhance delivery of O2 and glucose to and remove waste metabolites from hyper-metabolic brain regions.
As glioma cells migrate along the vasculature, the native cytoarchitecture is disrupted, opening the blood-brain barrier. Implantation of C6 rat glioma cells into the cerebrum of rats demonstrated that the processes and cell bodies of migrating glioma cells intercalate between vascular endothelial cells and astrocytic endfeet (Farin et al. 2006) . Tumor cells actively “lift up” astrocytic processes from the vasculature as revealed by transmission electron microscopy (Zagzag et al. 2000) . After movement of the astrocytic processes, glioma cells can directly attach to the perivascular basement membrane and cause the perturbed astrocytes to become “reactive” (Nagano et al. 1993) , which in turn can lead to downstream ramifications including loss of functional hyperemia and impairment of neuronal function . Incidentally, the association of glioma cells with the vasculature also seems to promote proliferation; more than 60 % of glioma cells that divided were less than 20 µm from a vascular branch point (Farin et al. 2006) .
14.2.2 Glioma Ion Channels Promote Aggressive Migration and Proliferation
As glioma cells migrate along the vasculature, they must move through the narrow extracellular spaces of the brain. To fit through these constrained spaces, glioma cells must undergo coordinated shape and volume changes, which are facilitated by ion channels and transporters expressed by glioma cells. Ion channels and transporters promote these morphological changes by moving osmotically-active ions across the plasma membrane, thereby osmotically driving water into and out of the cell. Given that approximately 70 % of the mass of a cell is water, this leads to robust changes in cellular volume, allowing glioma cells to navigate the tortuous extracellular environment of the brain.
Analogously, ion channel-mediated morphological changes are also important for glioma cell proliferation. The division of glioma cells is preceded by a short period of volume contraction known as pre-mitotic condensation . Pre-mitotic condensation is associated with the cell rounding and DNA condensation that occurs in a mother cell before dividing into two daughter cells (Habela et al. 2008) . This volume condensation is necessary for cell division, as its inhibition leads to an increase in glioma cell senescence.
14.2.2.1 Ion Channels Promote Hydrodynamic Shape and Volume Changes in Migrating Cells
Migrating human glioma cells shrink in volume by 33 % to move through narrow extracellular spaces irrespective of the size of the cell or spatial constraint (Watkins and Sontheimer 2011) . This dramatic decrease in cellular volume is accomplished through the efflux of Cl− ions into the extracellular space, leading to the obligated release of cytoplasmic water (Watkins and Sontheimer 2011) . Glioma cells accumulate large concentrations of Cl−, up to 100 mM, in the cytoplasmic space (Habela et al. 2009), likely due to the activity of NKCC1, a Cl− cotransporter highly expressed by human glioma cells (Haas and Sontheimer 2010) . The high intracellular [Cl−]i provides a large driving force for Cl− efflux, thereby moving water across the plasma membrane.
ClC-3 , a voltage-gated Cl− channel, has been identified as a key mediator of Cl− efflux by human glioma cells. ClC-3 is highly expressed on the plasma membrane (Olsen et al. 2003) and is activated by Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Huang et al. 2001; Cuddapah and Sontheimer 2010) , a Ca2+-sensitive kinase. CaMKII-dependent activation of ClC-3 is critical for glioma cell migration , as its inhibition either though pharmacological or genetic manipulation prevents glioma cell movement through spatial barriers (Cuddapah and Sontheimer 2010; Cuddapah et al. 2013a) . ClC-3 mediated Cl− flux is coupled to movement of K+ into the extracellular space through 2 Ca2+-activated K+ channels: KCa3.1 and KCa1.1 (Cuddapah et al. 2013a; Weaver et al. 2006) . Coupling of Cl− to K+ movement may allow migrating cells to maintain electroneutrality.
Intracellular [Ca2+] in gliomas increases secondary to a variety of ligands and channels, including glutamate , bradykinin , epidermal growth factor (EGF), lysophosphatidic acid (LPA), or transient receptor potential (TRP) channels, all of which enhance migration and invasion (Lyons et al. 2007; Ishiuchi et al. 2002; Montana and Sontheimer 2011; Bomben et al. 2011; Cuddapah et al. 2013a, 2013b; Manning et al. 2000) . Once Ca2+ increases, ion channels as well as cytoskeletal elements are regulated to promote migration. Specifically, three integral components of migration, including protrusion, attachment, and retraction, are activated. Protrusion involves the lamellipodium, rich with polymerizing actin filaments, which pushes the plasma membrane forward (Fig. 14.3a, 14.3b). Through a well-established treadmilling model, actin monomers away from the lamellipodium are depolymerized and actin monomers in the lamellipoium are polymerized, pushing the leading edge forward. This highly coordinated process requires Ca2+ signaling; Ca2+ signaling acts on Rho GTPases leading to downstream regulation of actin depolymerizing factor/cofilin family of proteins (Zheng and Poo 2007) . Additionally, focal adhesions, which bind migrating cells to the substratum and provide contact points to build traction, are regulated by CaMKII (Easley et al. 2008) . Thus Ca2+ dynamics are involved in the protrusion of the lamellipodium and attachment to the substratum (Fig. 14.3b). Importantly, increases in intracellular [Ca2+] also activate ion channels, which flux osmotically-active ions to move cytoplasmic water across the plasma membrane (Fig. 14.3c), allowing the motile cell to change shape and volume. For example, bradykinin-mediated Ca2+ elevations can lead to KCa 3.1 and CaMKII-dependent ClC-3 channel activation in human glioma cells (Cuddapah et al. 2013a) . This ion channel activation leads to shape and volume changes, and if inhibited, blocks migration (Watkins and Sontheimer 2011) . At the lagging edge myosin II contracts pushing the cell forward (Yang and Huang 2005; Conrad et al. 1993) (Fig. 14.3d). Given that a variety of ligands enhance glioma cell migration through an increase in Ca2+, this may be a convergent pathway common to multiple upstream signaling mechanisms.
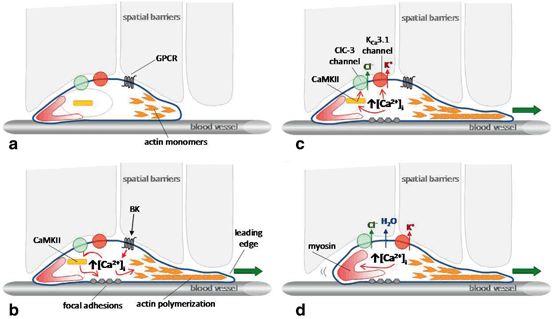
Fig. 14.3
An updated model of cell migration: Ion channels facilitate motility. a Glioma cells migrate along the abluminal surface of blood vessels and encounter spatial barriers requiring modulation of cellular shape and volume. b Ligands such as bradykinin can bind to G-protein coupled receptors (GPCRs) to increase [Ca2+]i, leading to CaMKII-mediated regulation of focal adhesions and Ca2+-mediated regulation of actin depolymerization via the actin depolymerizing factor/cofilin family of proteins. Actin polymerization on the leading edge pushes the leading edge forward. c [Ca2+]i increases also activate CaMKII-dependent ClC-3 channels leading to Cl- efflux, and KCa3.1 channels leading to K+ efflux. d Efflux of osmotically-active K+ and Cl- ions leads to loss of cytoplasmic water and cell shrinkage, facilitating movement through narrow spaces. Additionally, Ca2+-dependent myosin contraction on the lagging edge pushes the cell forward
14.2.2.2 Pre-mitotic Condensation is Necessary for Glioma Cell Proliferation
Similar to glioma cell migration , the division of glioma cells requires coordinated shape and volume changes. As glioma cells enter mitosis and prepare to divide into two daughter cells, cellular shape changes from bipolar to round (Cuddapah et al. 2011; Habela and Sontheimer 2007; Habela et al. 2008). The importance of a round morphology seems manifold, including allowing the cellular membrane and organelles to split symmetrically, and stabilizing the mitotic spindle for equal distribution of genomic content (Rosenblatt 2008) . This cellular rounding is associated with a simultaneous decrease in cytoplasmic volume by approximately 40 % termed pre-mitotic condensation (Habela and Sontheimer 2007; Habela et al. 2009). Importantly, CaMKII activation of ClC-3 has been identified as a mechanism that promotes pre-mitotic condensation in human glioma cells. Dividing glioma cells have a large Cl− conductance mediated by CaMKII activation of ClC-3 (Habela et al. 2008; Cuddapah et al. 2011). As in the context of migration, the large Cl− conductance in dividing human glioma cells is due to efflux of osmotically-active Cl− ions, which is sufficient to condense cytoplasmic volume by 33 % (Habela et al. 2009). Inhibition of ClC-3 or CaMKII significantly increases the time required for cytoplasmic condensation, thereby decreasing glioma cell proliferation by 35 % (Cuddapah et al. 2011).
This hydrodynamic process of pre-mitotic condensation may be coupled to other Ca2+-dependent processes to ensure robust proliferation in human glioma cells. During the first phase of mitosis, prophase, cyclin B binds to Cdk1 to promote cell division (Fig. 14.4a). The replicated chromosomes then begin to condense inside an intact nuclear envelope. Additionally, the replicated centrosomes begin to migrate to opposite poles of the cell. Between prophase and metaphase is prometaphase, which includes several steps preceding formation of the metaphase plate (Fig. 14.4b). The nuclear envelope breaks down, and the replicated chromosomes attach to microtubules through their kinetochores. The microtubules, in turn, are attached to centrosomes, forming the mitotic spindle. Ca2+ and calmodulin levels increase around the centrosome and microtubules (Welsh et al. 1979; Wolniak et al. 1983) to stabilize tubulin polymerization and depolymerization. As the cell transitions into metaphase, both centrosomes, already located at opposite poles of the cell, pull on the chromosomes (Fig. 14.4c). This tension generated by the opposing centrosomes pulls the chromosomes to the center of the cell forming the metaphase plate. The symmetric microtubule tension also drastically changes the cytoarchitecture leading to cellular shape changes. The metaphase cell begins to become round characterizing the process known as mitotic cell rounding. Towards the end of metaphase, intracellular [Ca2+] spikes for about 20 s (Ratan et al. 1988; Poenie et al. 1986) , potentially activating CaMKII and ClC-3 on the plasma membrane. As detailed above, because there is a large driving force for Cl− efflux (Habela et al. 2009), opening of ClC-3 channels leads to Cl− loss, subsequently driving the osmotic loss of cytoplasmic water. This cytoplasmic condensation leads to a shrinking of cellular volume, bridging kinases with their substrate proteins. After the intracellular [Ca2+] spike returns to baseline, daughter chromatids are separate to opposite ends of the cell towards the centrosomes, characterizing anaphase (Fig. 14.4d). As during prometaphase, Ca2+ and calmodulin levels remain elevated around the mitotic spindle, facilitating tubulin polymerization and depolymerization. This model suggests that Ca2+ is a master regulator and drives several necessary events between the metaphase-anaphase transition, and if disrupted, hinders mitotic progression. Ion channels play a critical role in mitosis by facilitating the dynamic shape and volume changes characteristic of dividing cells.
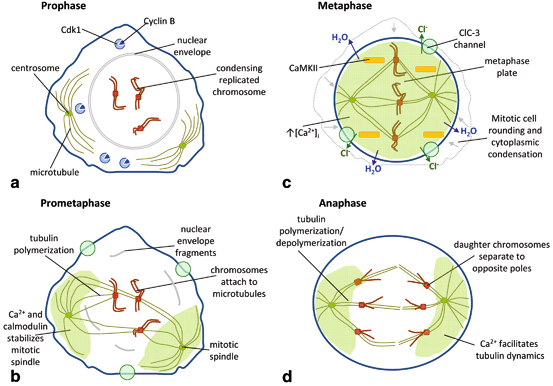
Fig. 14.4
An updated model of cell proliferation: Ion channels facilitate mitosis. a During prophase, cyclin B binds and activates Cdk1. The nuclear envelope is still intact, and the duplicated centrosomes begin to migrate to opposite ends of the cell. Replicated chromosomes begin to condense. b During prometaphase the nuclear membrane disintegrates, allowing microtubules to connect to kinetochores on the condensed chromosomes. On the other end, microtubules are attached to centrosomes, forming the mitotic spindle. Ca2+ and calmodulin stabilize tubulin polymerization on the mitotic spindle. c The dividing cell undergoes dramatic morphological changes during metaphase. The chromosomes align along the metaphase plate due to tension built by the mitotic spindle. The cell undergoes mitotic cell rounding. At the end of metaphase, [Ca2+]i spikes, potentially leading to activation of CaMKII then ClC-3. Activation of ClC-3 leads to Cl- efflux, osmotic water loss, and cytoplasmic condensation. d By anaphase, the daughter chromosomes are migrating to opposite poles of the cell. Once again, Ca2+ and calmodulin promote tubulin polymerization/depolymerization in the mitotic spindle
14.2.3 Glutamate: Motogen, Mitogen, and Neuron Killer
An important feature that distinguishes gliomas from other solid cancers is that tumor growth occurs in an unexpandable space, a space confined by the cranium. To overcome this spatial constraint, human glioma cells kill native brain tissue to create room for tumor expansion. One of the principal mechanisms by which glioma cells kill neurons is by release of large amounts of glutamate , the principal excitatory neurotransmitter of the nervous system. Glioma cells can increase the extracellularconcentration of glutamate to 100 µM in a space 1,000-fold larger than cellular volume within 5 h (Ye and Sontheimer 1999) . This is due to (1) a deficiency in glutamate uptake by glioma cells and (2) glutamate release by system xc- , a cystine-glutamate exchanger (Ye et al. 1999). Large extracellular glutamate concentrations in turn induce hyperexcitability and even excitotoxicity in neurons, causing neuronal death. Glutamate also acts in an autocrine manner, promoting glioma migration and growth by binding to Ca2+-permeable AMPA receptors/channels expressed by human gliomas.
14.2.3.1 Glutamate as a Motogen and Mitogen
One of the key functions of astrocytes is to take up glutamate from the extracellular space to dampen neuronal excitability. This glutamate uptake is primarily achieved through plasma membrane Na+-dependent glutamate transporters including GLT-1 and GLAST (in rodents; in human, EAAT2 and EAAT1, respectively). In contrast, gliomas, the malignant counterpart of astrocytes, fail to take up glutamate from the extracellular space (Ye and Sontheimer 1999) . Na+-dependent glutamate uptake is up to 100-times less in human glioma cells, secondary to decreased GLT-1 expression and mislocalization of GLAST to the nucleus (Ye et al. 1999) . Instead, glutamate is persistently released through the system xc- cystine-glutamate exchanger. Cystine enters the cell and is converted to glutathione, an antioxidant critical for the absorption of free radicals formed by gliomas in hypoxic environments (Ogunrinu and Sontheimer 2010) . Glutamate released in the extracellular space (1) acts in an autocrine loop on gliomas to increase tumor aggressiveness, and (2) induces hyperexcitability in peritumoral neurons and eventual excitotoxic cell death.
Human glioma cells lack the GluA2 subunit in endogenously expressed AMPA receptors, rendering the channels Ca2+-permeable (Ishiuchi et al. 2002, 2007; Lyons et al. 2007) . Therefore, when glutamate is released from the system xc-, it then acts in an autocrine/paracrine manner to increase Ca2+ signaling in glioma cells, which correlates with increased migration (Ishiuchi et al. 2002; Lyons et al. 2007) . Inhibition of glutamate release from the system xc- or inhibition of Ca2+-permeable AMPA receptors decreased glioma cell migration and reduced overall tumor growth (Lyons et al. 2007). Additionally, conversion of AMPA channels from Ca2+-permeable to Ca2+-impermeable by overexpression of the GluA2 subunit significantly decreased tumor volume by inhibiting glioma proliferation and invasiveness and increasing glioma cell apoptosis (Ishiuchi et al. 2002). Mechanistically, Ca2+ permeation through AMPA receptors may promote proliferation and migration through the activation of Ca2+-sensitive ion channels that facilitate hydrodynamic shape and volume changes (Cuddapah et al. 2011, 2013a) . Alternatively, Ca2+ influx may activate Akt, thereby enhancing signaling cascades that lead to proliferation and migration (Ishiuchi et al. 2007).
14.2.3.2 2Gliomas Induce Seizures and Excitotoxicity Through Release of Glutamate
Beyond autocrine/paracrine signaling, glioma-released glutamate acts on neurons to induce hyperexcitability and eventual neuronal death. Human gliomas implanted into mouse cerebrums induced epileptic activity within 14–18 days, as measurable by electroencephalogram (Buckingham et al. 2011) . This hyperexcitability was due to glutamate release from the system xc- acting at peritumoral neurons (Buckingham et al. 2011). Importantly, from a clinical perspective, pharmacological inhibition of the system xc- reduced neuronal hyperexcitability and seizure activity (Buckingham et al. 2011; Campbell et al. 2012) .
As extracellular glutamate concentrations increase in the peritumoral environment, neuronal hyperexcitability snowballs into excitotoxicity . Abnormal glutamate activation of ionotropic glutamate receptors leads to excessive Ca2+ influx, leading to the activation of phospholipases and proteases associated with excitotoxicity. Incubation of neurons in media exposed to human glioma cells is sufficient to cause sustained Ca2+ increases in neurons, characteristic of excitotoxicity (Ye and Sontheimer 1999) . This leads to neuronal death and can be reversed by blocking Ca2+-permeable N-methyl-D-aspartate (glutamate) receptors with MK-801 or by inhibiting glutamate release from glioma cells (Ye and Sontheimer 1999).
14.2.4 Malignant Gliomas Are a Neurodegenerative Disease
As detailed above, glutamate release through the system xc- by gliomas leads to excitotoxicity and neuronal death. Neuronal death is a hallmark of neurodegeneration, an umbrella term for diseases that lead to the malfunctioning or loss of neurons. The term “neurodegeneration” is typically applied to Alzheimer’s disease, Parkinson’s disease , Huntington’s disease , amyotrophic lateral sclerosis (ALS) , and trauma/injury to the central nervous system , all diseases which involve a progressive decrease in neuron and synapse counts. While the cause of neuronal loss can be variable, including protein misfolding, protein aggregates , astrogliosis , or loss of antioxidants, the endpoint is the same. Given that malignant gliomas lead to progressive neuronal loss, it too can be considered as a neurodegenerative disease. Appreciating gliomas as a form of neurodegeneration fosters more of a neurocentric approach. Approaching gliomas from a purely oncological perspective, as has historically been the case, does not take into account the unique brain microenvironment and its effects on disease progression.
14.2.4.1 Vasculature Disruption in Neurodegeneration
Cerebral vascular abnormalities typify many neurodegenerative diseases , and malignant gliomas certainly meet this criterion. Neurodegeneration, including neuronal loss and deficits in memory and learning, can directly result from poor microvascular circulation secondary to ablation of pericytes, an important component of the neurovascular unit (Bell et al. 2010) . Additionally, mutations in superoxide dismutase 1 (SOD1), which can lead to ALS, cause disruptions in the blood-spinal cord barrier by inducing downregulation of several endothelial tight junction proteins including ZO-1 and occludin (Zhong et al. 2008) . Interestingly, the breakdown of the blood-spinal cord barrier precedes motor neuron loss in the ventral horn that is typical of the disease (Zhong et al. 2008) , suggesting the vascular breakdown plays a causative role in neurodegeneration. Alzheimer’s disease is also associated with disrupted vascular tone as amyloid beta deposits lead to disorganization of vascular smooth muscle cells (Christie et al, 2001). When gliomas are implanted in to mice overexpressing amyloid beta, tumors are less vascularized, demonstrating the inherent antiangiogenic properties of amyloid beta (Paris et al. 2010) . Similar to other neurodegenerative conditions, gliomas are characterized by a malfunctioning blood-brain barrier and poor vascular perfusion. Glioma cells migrate along the vasculature, disrupting native cytoarchitecture, and induce angiogenesis, leading to the growth of leaky and hypo-functional vessels. Dysfunctional vessels may lead to a loss of coupling between neuronal firing and vascular perfusion and yield focal symptoms associated with gliomas.
Related posts:
 General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
 Ionic Signaling in Physiology and Pathophysiology of Astroglia
Ionic Signaling in Physiology and Pathophysiology of Astroglia
 Amyotrophic Lateral Sclerosis: A Glial Perspective
Amyotrophic Lateral Sclerosis: A Glial Perspective
 Enteric Glial Cells: Implications in Gut Pathology
Enteric Glial Cells: Implications in Gut Pathology
 Microglial Biology in Neuroinflammatory Disease: Pharmaco-industrial Approach to Target Validation
Microglial Biology in Neuroinflammatory Disease: Pharmaco-industrial Approach to Target Validation
 Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


