Chapter 10 Orbit
OCULAR LESIONS
Computed tomography (CT) is somewhat limited in the diagnosis of ocular lesions because of its present inability to resolve the retina, choroid, and sclera. It is exquisitely sensitive to ocular calcification. Box 10-1 lists the causes of ocular calcification (Fig. 10-1). On T1-weighted images (T1WI) and T2-weighted images (T2WI), most pure calcifications (without paramagnetic ions) are hypointense and do not change in intensity as one increases the time to echo (TE). This is caused by the lack of mobile protons. In some instances, calcification is of high intensity on T1WI. Most of these situations occur in the basal ganglia but can also occur with ocular calcifications, as in retinoblastoma. Gradient-echo imaging sequences can be employed that emphasize T2* effects (see Chapter 1) and hence increase the conspicuity of ferrocalcinosis. In this case, susceptibility differences produce hypointensity so that calcium, which has altered diamagnetic susceptibility compared with tissue, would appear hypointense. This is sometimes useful in detecting calcium on a magnetic resonance (MR) scan of a patient with retinoblastoma. This hypointensity is not specific for calcium; other entities, including blood products (deoxyhemoglobin, intracellular methemoglobin, hemosiderin) and paramagnetic compounds (including the melanin in melanoma), may be hypointense on gradient-echo images.
Vascular
Persistent Hyperplastic Primary Vitreous
Persistent hyperplastic primary vitreous is caused by persistence of various portions of the primary vitreous (embryonic hyaloid vascular system) with hyperplasia of the associated embryonic connective tissue and may present as leukokoria in the neonate. The eye usually is microphthalmic, although the degree of microphthalmia can be minimal, and the lens can be small with flattening of the anterior chamber. Initially, a funnel-shaped mass of fibrovascular tissue (including the persistent hyaloid artery) is present in the retrolental space and runs in an S-shaped course (termed Cloquet’s canal) between the back of the lens and the optic nerve head. PHPV is vascular and prone to repeated hemorrhages and may vary in size. CT demonstrates generalized increased density in the globe and enhancement of the intravitreal tissue after intravenous contrast. Unlike retinoblastoma, PHPV does not calcify (this is key!). The CT diagnosis is facilitated by observing the S-shaped tubular fetal tissue between the lens and the optic nerve head (Fig. 10-2). A blood-vitreous layer may be seen as a result of posterior hyaloid detachment. On MR, vitreal intensity is variable; however, the visualization of Cloquet’s canal makes the diagnosis. This is an entity that may lead to high signal on T1WI in the vitreous, possibly from hemorrhage or high protein, with low or high signal on T2WI.
Infectious/Inflammatory
Cytomegalovirus
Cytomegalovirus (CMV) is the most common opportunistic infection of the retina and choroid. Up to one fourth of patients with AIDS have CMV infection. It is hemorrhagic and can produce high density on CT or various MR appearances depending on the stage of blood (Fig. 10-3). CMV retinitis can result in retinal detachment, the treatment of which may require intraocular injections of silicone oil or other high-density substances. Thus, ocular infection by CMV can lead to a myriad of imaging findings. Other less common infectious pathogens affecting the retina, choroid, or both in AIDS include Toxoplasma gondii, Mycobacterium tuberculosis, Pneumocystis carinii, and the herpesviruses.
Posterior Scleritis
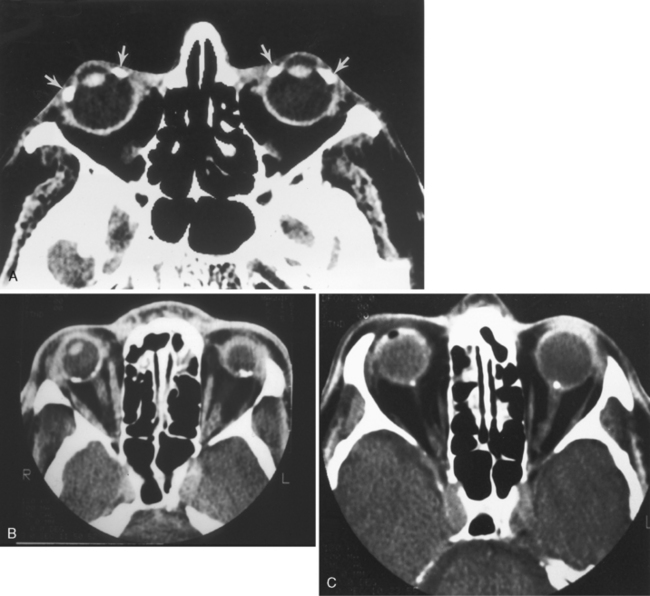
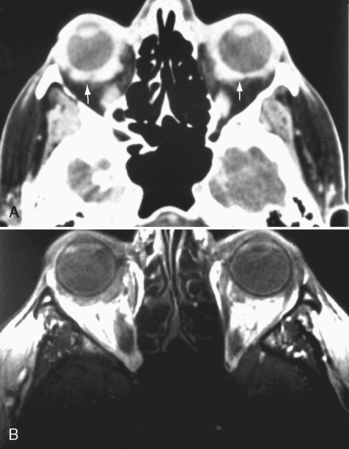
This can be infectious (bacterial, fungal, or viral) or autoimmune (collagen vascular diseases versus sarcoidosis) in origin. It is separated into acute and chronic categories. Findings include thickening and enhancement of the posterior sclera and uvea (Fig. 10-4). The thickening may be nodular or diffuse. The nodular variety can be mistaken for melanoma; the diffuse type can mimic lymphoma in its uniform thickening. The disease may be associated with retinal or choroidal detachments and uveitis.
Trauma
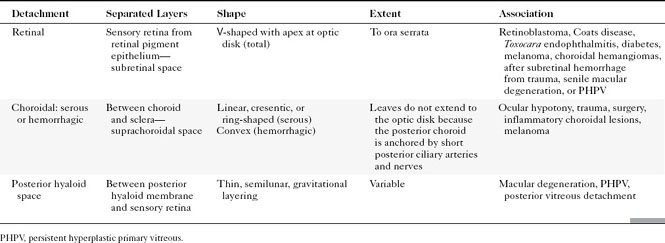
There are four potential spaces for “bad humor” to accrue between the membranes of the globe in trauma and in infection. The space between the base of the vitreous and the sensory retina is the posterior hyaloid space. The space between the layers of the retina (sensory retina and retinal pigment epithelium) is the subretinal space, and between the choroid and the sclera is the suprachoroidal space (Fig. 10-5). When the posterior hyaloid membrane separates from the sensory retina, the separation is termed a posterior vitreous detachment. It is curvilinear in shape, anterior to the retina, and separate from the optic disc. Total retinal detachments are V shaped, with the apex pointing to the optic disc and the arms at the ora serrata (the anterior aspect of the retina). In fact, retinal separation is probably a better term because the two layers of the retina—neurosensory and retinal pigment epithelium—separate. Choroidal detachments are generally limited by the vortex veins or posterior ciliary arteries and usually do not reach the optic disc. In addition, choroidal detachments can extend anteriorly beyond the ora serrata to the ciliary body, occasionally detaching it. Subtenon space is located between the sclera and the fibrous membrane (Tenon capsule) adjacent to the orbital fat, extending from the ciliary body to the optic nerve. Hemorrhages in this space, most often from trauma, conform to the curvilinear shape of the eyeball (Fig. 10-6).
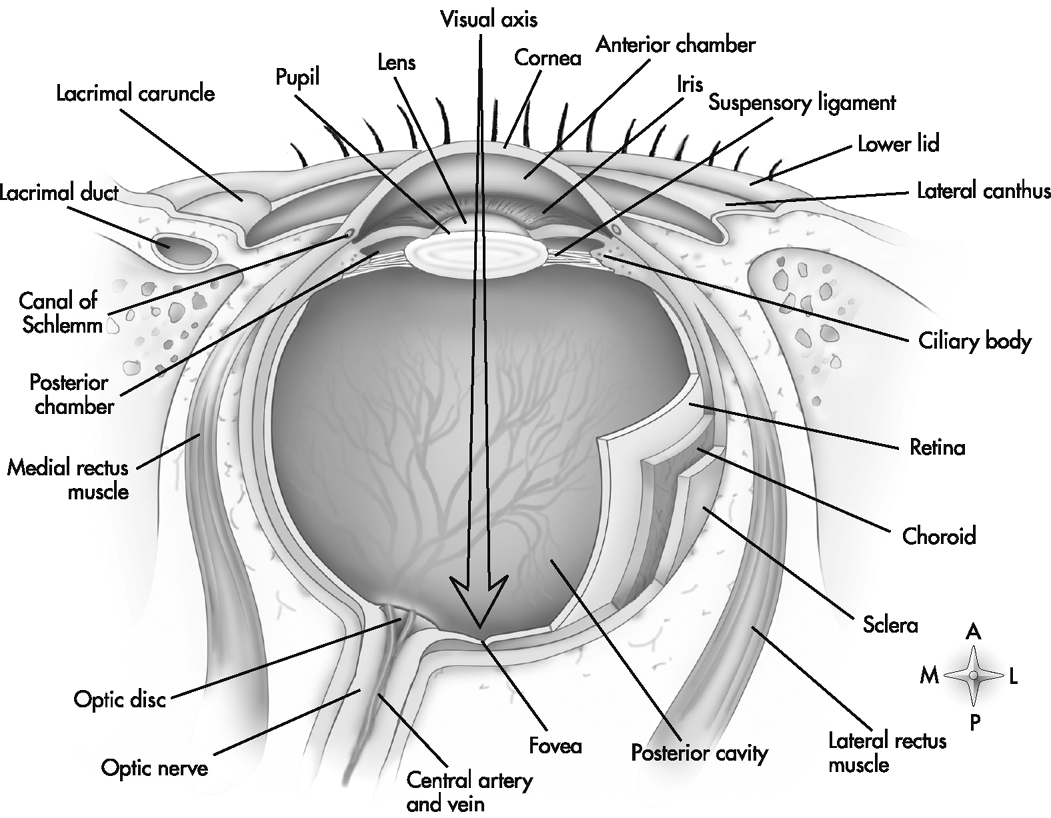
Figure 10-5 Horizontal section through left eyeball, viewed from above.
(From Thibodeau GA, Patton KT: Anatomy and Physiology, 4th ed. St. Louis, Mosby, 1999, p. 462. Used with permission.)
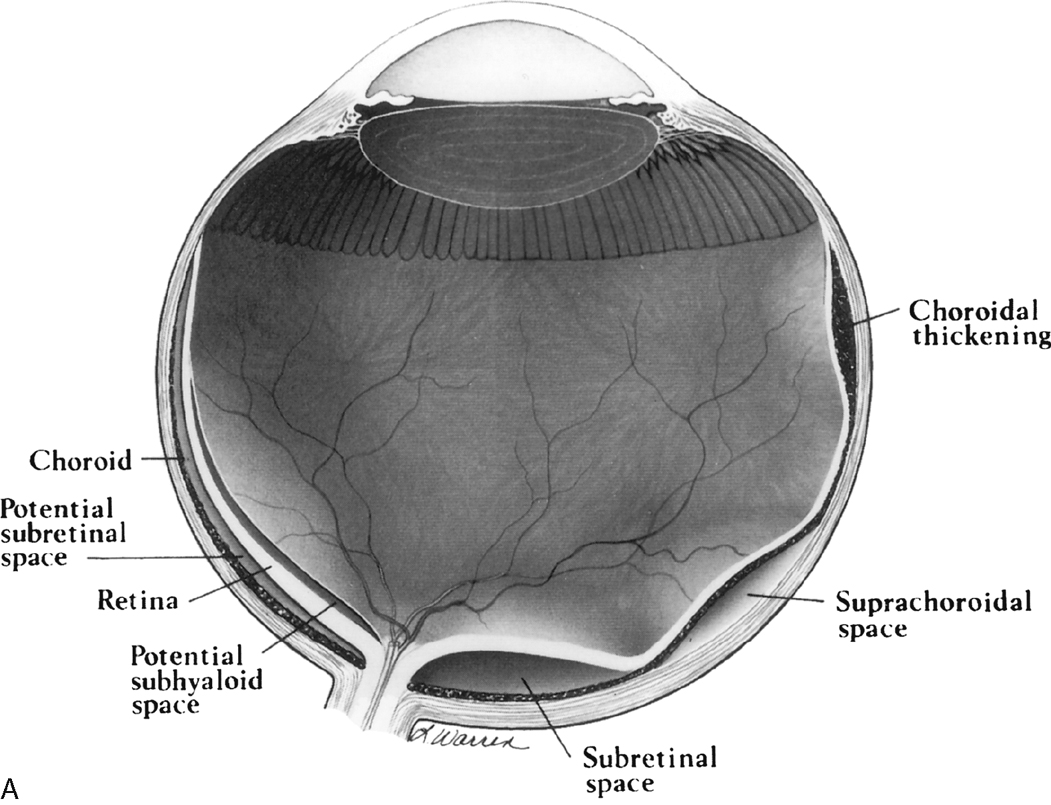
Ocular Hypotony and Choroidal Detachments
Ocular hypotony is defined as low intraocular pressure secondary to surgery, trauma, or glaucoma therapy and may be observed on CT as uveoscleral infolding, also referred to as the “flat tire” or “umbrella” sign (Fig. 10-7). This is most often seen after globe perforation and can be reversed by instillation of saline, silicone oil, or sodium hyaluronate (Healon), which expands the globe. Just two more words: double perforation. This term is used by ophthalmologists and means a perforation through the anterior and posterior portions of the globe. Suspected posterior perforation is one reason to perform an imaging study; it usually cannot be repaired (poor outcome). The most common cause of a double perforation is BB-sized shot.
Ocular hypotonia, from inflammatory diseases (uveitis or scleritis), medication, or traumatic perforation of the globe, is the cause of serous choroidal detachments. Table 10-1 compares the various ocular detachments radiologists commonly encounter (see Fig. 10-7). Hemorrhagic choroidal detachment may be observed after penetrating injury or intraocular surgery and has a lenticular morphology. This is bad news because it means that you have had a rupture of a choroidal vessel, which has an associated poor prognosis. Serous choroidal detachment (benign prognosis) is crescentic or ring-shaped, and the curvilinear choroid can be identified. Serous choroidal effusions beneath the detached choroid appear as convex regions of low density outlined by the detached choroid. Hemorrhagic choroidal effusions are high density and do not change with position. Inflammatory choroidal effusions secondary to uveitis and posterior scleritis are high density but may change in location with changes in head position. Uveoscleral thickening and enhancement can be seen in inflammatory choroidal detachment. MR is beneficial in distinguishing hemorrhagic choroidal detachment from serous choroidal detachments. On MR, hemorrhagic choroidal detachment can display a variety of intensity patterns related to age of the hemorrhage (see Chapter 4). Serous choroidal detachment has been noted to be high intensity on T1WI and T2WI.
Acquired and Metabolic Disorders
Drusen
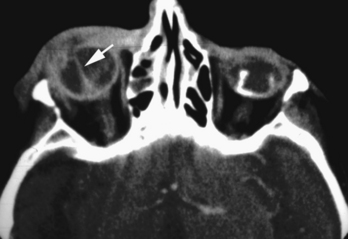
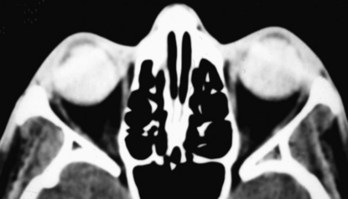
The most common calcifications in the globe are drusen. Optic nerve head (optic disk) drusen are composed of a mucoprotein matrix with significant quantities of acid mucopolysaccharides together with small quantities of ribonucleic acid and, occasionally, iron. They are laminated calcareous deposits located in the substance of the optic nerve anterior to the lamina cribrosa. This condition has an incidence of up to 2% of the population. Drusen may be familial (typically bilateral) or may be associated with other conditions, such as retinitis pigmentosa. They may be unilateral or bilateral and are located just anterior to the junction of the optic nerve and globe. CT demonstrates punctate calcifications in this region (see Fig. 10-1C). MR has difficulty detecting this condition because of its relative insensitivity to calcium. The clinical issue for the radiologist is that if the drusen are buried below the surface of the nerve head, they may not be seen by the ophthalmologist, and the patient may be referred for a workup of what appears to be papilledema (hence, drusen are a cause of pseudopapilledema). Rarely one might consider a differential diagnosis of retinoblastoma or astrocytic hamartoma in a child or adult with extensive optic disk calcification; however, drusen have no enhancement or soft-tissue mass on enhanced MR, as opposed to retinoblastoma.
Senile/Senescent Calcifications
As part of the aging process, fine pinpoint calcifications may develop at or just anterior to the insertion sites of the extraocular muscles on the globe within the sclerochoroidal layers. These areas can be appreciated on CT (see Fig. 10-1A). Hyperparathyroidism and hypomagnesemia predispose to these calcifications. Trochlear calcification may be symptomatic with pain and restricted movement as a tenosynovitis known as Brown syndrome, but it is more commonly asymptomatic and associated with diabetes mellitus.
Neoplastic
Retinoblastoma
Although ophthalmoscopy is usually able to visualize retinoblastoma, imaging is of critical importance in revealing retrobulbar extension of the tumor and invasion of the optic nerve posterior to the lamina cribrosa, where the meninges surround the optic nerve and are contiguous with the subarachnoid spaces of the central nervous system (associated with a poor prognosis) (Box 10-2).
Retinoblastoma may spread directly into the subarachnoid space via the optic nerve (occasionally producing spinal implants), hematogenously, or via lymphatics and has a propensity to hemorrhage. Metastases from retinoblastoma occur within the first 2 years after treatment. There is a high incidence of nonocular malignant tumors in patients with the hereditary form of retinoblastoma, leading to a 59% 35-year mortality rate. These include midline, primitive neuroectodermal tumors (PNETs), pineal tumors, osteogenic sarcoma, soft-tissue sarcoma, malignant melanoma, basal cell carcinoma, and rhabdomyosarcoma (Fig. 10-8). The pineal gland in lower animals contains photoreceptors and has been termed the “third eye,” so that the triad of a pineal tumor (usually a pineoblastoma) and bilateral retinoblastoma has been termed trilateral retinoblastoma (Fig. 10-9). The pineoblastoma may be detected simultaneously with, after, or even before the ocular lesion. Ectopic retinoblastoma has also been reported in the parasellar or suprasellar regions.
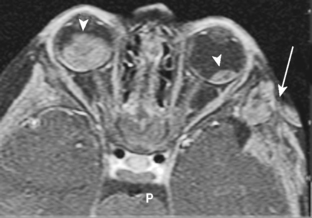
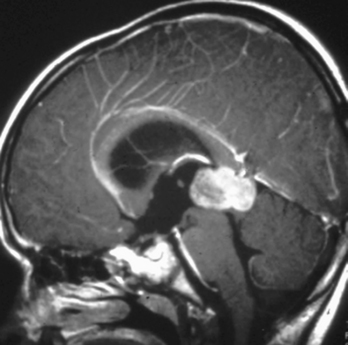
Figure 10-9 Trilateral retinoblastoma. Note the pineoblastoma in a patient with bilateral retinoblastoma.
The CT findings of retinoblastoma are calcifications in the posterior portion of the globe with extension into the vitreous (Fig. 10-10). The calcification may be homogeneous or irregular and occurs in 95% of patients. Noncalcified retinoblastoma has been reported in infants with a family history, under close observation. In children younger than age 3 years, ocular calcification is highly suggestive of retinoblastoma; conversely, its absence in this same group makes the diagnosis of retinoblastoma highly unlikely. These lesions minimally enhance; however, contrast is useful to appreciate intracranial extension. With orbital involvement, proptosis may be seen. Coronal imaging is useful in assessing enlargement of the optic nerve secondary to invasion by the tumor.
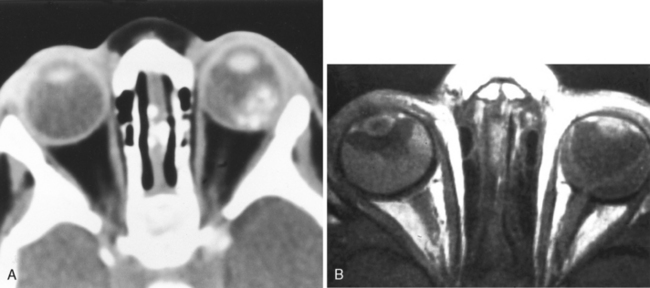
Magnetic resonance findings in retinoblastoma include moderately high intensity on T1WI (Fig. 10-10B) and hypointensity on T2WI, with the noncalcified portion of the lesion less hypointense than the calcification. We speculate that the high intensity on T1WI may be caused by calcium or by some coexistent paramagnetic, tumor protein, or hemorrhagic component. Associated retinal detachment may be high intensity on T1WI and of variable intensity on T2WI, depending on the protein content and whether there is associated hemorrhage in the subretinal fluid. Enhancement is often present and may be the only indicator of spread to the retrobulbar compartment or along the subarachnoid space surrounding the optic nerve, intracranial, or intraspinal spaces. In patients with suspected retinoblastoma CT and MR are complementary examinations. CT detects calcification better than MR, whereas MR is more sensitive than CT for tumoral extension into the nerve and intracranial space, and for detecting secondary lesions.
Melanoma
Melanomas of the uveal tract are the most common intraocular malignancy in adults and have a characteristic intensity pattern on MR. They are rare in African Americans but when they occur tend to be larger, more pigmented, and more necrotic. Conditions predisposing to uveal melanoma include congenital melanosis, ocular melanocytosis, oculodermal melanocytosis, and uveal nevi. Unlike most other tumors, melanotic melanomas are hyperintense on T1WI and hypointense on T2WI. Free radicals known to exist in melanin are responsible for the associated T1- and T2-shortening by the proton-electron dipole-dipole proton relaxation enhancement mechanism (Fig. 10-11). The degree of T1- and T2-shortening appears to be related to the melanin content. Amelanotic melanomas have MR characteristics similar to those of other tumors (hypointense on T1WI and hyperintense on PDWI/T2WI). Other lesions with similar intensity patterns that could be confused with melanotic melanoma include fat and subacute hemorrhage in the intracellular methemoglobin state. Use of fat saturation images can easily separate fat from melanotic tumor. There have been reports of mucin-secreting adenocarcinoma metastatic to the choroid and other carcinomas having similar signal characteristics to a choroidal melanotic melanoma. Associated with the melanoma may be a retinal detachment, with a subretinal proteinaceous effusion that appears as high intensity on T2WI. To differentiate melanoma from other lesions, T1WI, T2WI, and enhanced scans must be used.
Metastases
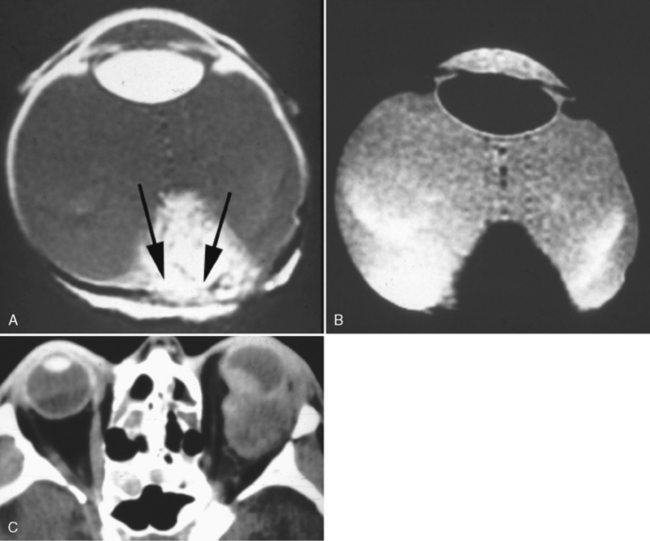
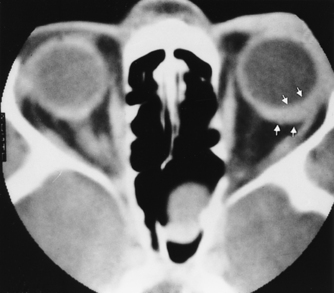
Metastatic breast and lung cancer, lymphoma, and leukemia are common ocular malignancies in adults. Many metastatic neoplasms of the globe produce eccentric thickening of the uveoscleral rim and retrobulbar extension that may not be clinically apparent but that can be detected on CT and MR (Fig. 10-12). Choroidal lymphoma and leukemia can appear similar to uveal melanoma on MR, may cause retinal detachment, and may be bilateral (rare for melanoma) (Fig. 10-13).
Congenital
Coloboma
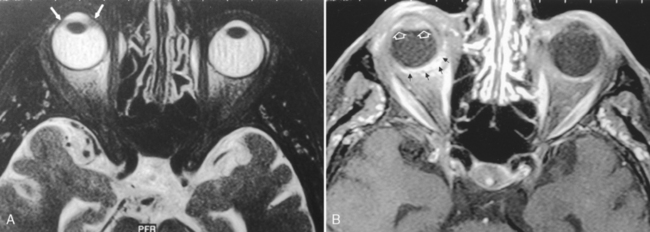
On imaging, there is a cone- or notch-shaped deformity, usually of the posterior globe, that may involve the optic nerve, with eversion of a portion of the posterior globe (Fig. 10-14). Other structures, including retina, choroid, iris, and lens, can be affected. The sclera is normal. Colobomas usually arise sporadically and are unilateral, although rarely an autosomal dominant form can exist, with about 60% of those affected having bilateral coloboma.
Phakomatoses
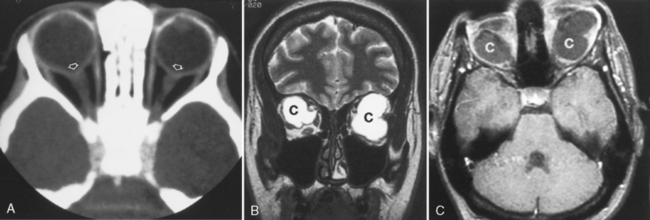
Retinal astrocytic (glial) hamartomas are observed in cases of tuberous sclerosis, neurofibromatosis and, rarely, von Hippel-Lindau disease (see Chapters 3 and 9). On CT, a focal region of calcification is noted in the retina that is much more extensive than that of optic disc drusen and can potentially be confused with retinoblastoma. These hamartomas grow slowly and do not metastasize. A hallmark of von Hippel-Lindau disease is retinal hemangioblastoma (which cannot be identified angiographically). Hemangioblastomas of the retrobulbar optic nerve and orbit have also been reported in von Hippel-Lindau disease. The primary differential diagnosis of such optic nerve lesions is angioblastic meningioma and optic nerve glioma. Half the patients with optic nerve hemangioblastoma have other associated lesions of von Hippel-Lindau disease. Choroidal hemangiomas are reported in Sturge-Weber syndrome (tomato ketchup retina).
Drug-related/Iatrogenic
Prosthetic Intraocular Lens
Contemporary cataract surgery usually includes placement of an intraocular lens. These lenses can be identified on CT or T2WI. This becomes an issue in orbital trauma when concern is raised regarding the position of the lens or about whether the patient has one. The crystalline lens can be dislocated in a variety of different medical conditions. Box 10-3 provides a list of these conditions as well as the common position of these dislocations.
The Big or Small Globe (Box 10-4)
Occasionally, one is confronted with an image of an enlarged globe. The most common cause of this is axial myopia. Other conditions causing enlarged globes include congenital glaucoma (buphthalmos), collagen disorders (e.g., Marfan syndrome), posterior staphyloma (Fig. 10-15), coloboma, juvenile glaucoma in patients with Sturge-Weber syndrome, proteus syndrome, and neurofibromatosis.
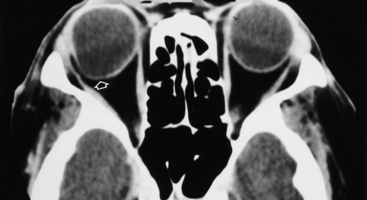
Small globes (microphthalmia) have been reported as an isolated event or may be associated with conditions such as craniofacial anomalies, congenital rubella, PHPV, ROP, and phthisis bulbi (a small shrunken calcified globe, usually the result of a traumatic injury) (Fig. 10-16). Anophthalmia represents complete absence of ocular tissue.
EXTRAOCULAR
Lesions in the orbit may be classified as being conal, intraconal (IC), extraconal (EC), or both, depending on their relationship to the muscle cone in the orbit. For the purposes of the extraocular compartment, we review the anatomy briefly and then describe the pathology using the same categories as used previously. After each entity’s header we use the abbreviations of IC or EC to denote in which compartment the lesion is more common. The differential diagnosis of lesions by virtue of their anatomic origin is provided in Boxes 10-5 to 10-8. MR, by virtue of its superior anatomic and multiplanar imaging, is the imaging modality of choice for localizing orbital lesions.









