7 As would be expected in a disorder caused by the loss of a tumor suppressor gene, neurofibromatosis 1 (NF1) increases the risk of developing multiple types of tumors in both the central and peripheral nervous systems. Although most NF1-associated tumors are benign neurofibromas (see Chapter 6), the disorder also increases the risk of other types of tumors and certain malignancies. The tumors that arise in the central nervous system (CNS) are potentially more severe in terms of medical impact than are neurofibromas. The CNS tumors most likely to occur in NF1 are optic glioma (a tumor of the optic nerve) and astrocytoma (a type of brain tumor). People with NF1 also develop pheochromocytoma (an adrenal gland tumor), juvenile myelomonocytic leukemia (a blood cancer), and rhabdomyosarcoma (a muscle cell tumor) more frequently than the general population. Because these cancers are all uncommon, the overall risk for someone with NF1 is still low.1 In addition, NF1 exerts multiple effects on the developing nervous system, in ways that researchers are only beginning to understand. The most common nervous system abnormalities seen in NF1 are macrocephaly (a large head) and headaches. Less frequently, epilepsy and hydrocephalus (fluid buildup in the brain) may occur. This chapter discusses the most common CNS tumors and other malignancies in NF1 and nervous system abnormalities typical of the disorder. Optic pathway gliomas are tumors that develop from cells known as astrocytes that surround the optic nerve. The tumors are called gliomas because astrocytes are among the most common glial cells in the brain (see Chapter 3). Optic gliomas are common in people with NF1, present in an estimated 15% of people with the disorder, although there is evidence that these tumors occur less frequently in African Americans.2–4 In most cases, however, the tumors are slow growing, do not cause symptoms, and do not require treatment. Only 5% of children with NF1 develop vision abnormalities related to an optic glioma, for instance.2 Some children with an optic glioma may experience early puberty, depending on the tumor’s location. Optic pathway gliomas develop in the preschool years, with most presenting between the ages of 4 and 6. For that reason, an annual eye examination by an ophthalmologist is recommended for any child with NF1.1 Although other types of routine screening, such as magnetic resonance imaging (MRI) scans, are not recommended unless symptoms develop, this remains a controversial issue. Some physicians advocate routine MRI scans to determine whether or not a child with NF1 has developed an optic glioma. The consensus remains, however, that neuroimaging studies may detect asymptomatic tumors, but they do not prevent symptoms from developing, improve treatment, or alter longterm outcomes.5 Optic gliomas are low-grade tumors and are usually not aggressive.6(p235) They can develop anywhere along the optic nerve, including inside the eye socket (the intraorbital portion of the nerve), inside the skull (intracranial portion), on the optic chiasm (the area in the brain where the optic nerve from each eye crosses before extending further into the brain), or on the optic tracts leading to the visual cortex. Most optic gliomas are found in the intraorbital and intracranial sections of the optic nerve or the optic chiasm.6(p205) Although optic pathway gliomas constitute only 2 to 5% of all childhood brain tumors, children with NF1 have 70% of those that are diagnosed.6(p203) It is not clear why optic pathway gliomas occur more often in people with NF1 than in the general population. Analysis of autopsied tissue from people, as well as laboratory studies in mice, indicates that the NF1 gene mutation, which causes a loss of neurofibromin protein, somehow encourages an increase in the proliferation of astrocytes. The overabundance of these cells contributes to tumor formation (see Chapter 3). The tumors that progress and cause symptoms quite likely do so because of the influence of hormones and/or of additional genetic mutations.6(p207) This theory is bolstered by population-based (epidemiological) studies that have found that in children with NF1 girls are twice as likely to develop an optic glioma as boys.7,8 As many as two thirds4 of optic gliomas do not cause symptoms. About half6(p208) of people who do become symptomatic experience vision abnormalities, but these rarely get worse and can either go without treatment or be managed with eyeglasses. In one in three people with symptomatic tumors, the tumor causes progressive proptosis,8–10 in which the eye bulges outward. Complete or partial loss of vision may result. Another potential complication of optic pathway glioma is premature puberty, which results in about one in three children who develop symptoms.8 Precocious puberty is that which occurs before the age of 7 in girls and 9 in boys.6(p215) Early puberty occurs when the optic glioma is located in the optic chiasm and places pressure on the hypothalamus, the area of the brain that controls the secretion of hormones, among other functions. The first sign that a child with NF1 may experience precocious puberty is a sudden growth spurt, so that the child is taller than the normal range for peers (see Chapter 9). Regular vision examinations help to detect symptoms indicating the presence of an optic glioma. A child with NF1 should be examined by a pediatric ophthalmologist or an ophthalmologist experienced in treating people with NF1. The examination should include an assessment of visual acuity, color vision, and visual fields, as well as examination of the optic nerve and eyeball. Annual visits are recommended for all children with NF1 until 10 years of age,1 and afterward if an optic glioma or vision irregularity is detected. Asymptomatic older children and adults should also undergo regular eye exams, although how frequently they occur and how extensive they should be may vary. (Ask an NF specialist for personal advice.) Any of the following symptoms or clinical findings made during an ophthalmological exam may indicate the presence of an optic glioma. However, a normal exam does not rule out the presence of a tumor. Two of three people with an optic glioma have normal vision and other findings on an eye exam.11(p85) Performance on a vision test indicates whether there is vision loss in one or both eyes. This can be a difficult screening test for young children, yet that is the age when the optic glioma is most likely to develop. This is another good indication that a tumor may be present on the optic nerve, but it may be difficult to assess in young children. When a light is shined suddenly at a normal eye, pupils in both eyes constrict. In an eye with optic nerve damage, however, the pupil may dilate—an indication that the nerve is not functioning properly and the information it conveys to the brain may be garbled. Defects in the visual field include deterioration of the central or peripheral vision and “blind” spots. Visual field testing is hard to do in children under age 7, those most likely to harbor these defects. Experts recommend that this type of test be reserved for children older than 7 who are known to have lesions in the optic chiasm. In this case, visual defects will be present in both eyes.6(p205) This is a swelling of the optic nerve caused by increased pressure inside the skull. When the nerve atrophies, it loses color and mass. Inasmuch as this manifestation occurs after others have become evident, it may not be useful in diagnosis. This test measures electrical activity in the brain’s visual cortex to gauge the health of the visual pathways. (The patient watches a monitor with moving patterns while electrodes are attached to the scalp.) Although this test may be useful in evaluating the health of the optic nerve, it does not always reveal the presence of an optic glioma. Many people with such tumors perform normally in the visual evoked response test. If an eye examination or visual evoked response test reveals telltale vision discrepancies or optic nerve dysfunction, an MRI of the head and eye sockets will reveal whether a tumor is growing along the optic nerve (Fig. 7–1). If the child does have an optic glioma, monitoring should consist of regular follow-up ophthalmological exams, to determine whether symptoms are getting worse, and periodic MRIs as recommended by a physician. Figure 7–1 Magnetic resonance imaging (MRI) reveals an optic glioma of the left optic nerve (arrow). All children with NF1 should have their height and weight measured at least annually and compared with standard growth charts. The first sign that the child will experience premature puberty is accelerated growth in height. This may also be the first sign that the child has an optic glioma, even when vision tests are normal. Because most optic gliomas do not progress, become invasive, or cause any symptoms,3 the best strategy is one of watchful waiting and regular follow-up eye exams. A small proportion of optic pathway gliomas require treatment because they compromise vision, are disfiguring, or compress adjacent brain structures.12 Because of the location of these tumors, surgical resection is difficult. The only way to completely remove the tumor is to remove the optic nerve, which would cause blindness in that eye. Sometimes surgery is useful in reducing the size of the tumor (a process known as debulking it). The mainstay of treatment is chemotherapy, usually with a combination of vincristine and carboplatin, although carboplatin may be used alone.5,13 If necessary, this initial treatment is followed by radiation, usually when the child is older to minimize long-term side effects. The goal of treatment is to reduce the size of the tumor, retard further tumor growth, and preserve vision. A risk of chemotherapy and radiation is that secondary malignancies may develop, as they sometimes do in other disorders involving loss of a tumor suppressor gene. Radiation may damage blood vessels (see the discussion on moyamoya in the subsection Vasculopathy in Chapter 9), leading to stroke, and may contribute to cognitive difficulties. It may interfere with the brain’s ability to produce certain hormones, thereby interfering with the child’s growth and development.11(p86) People with NF1 may develop other types of astrocytomas, although these other brain tumors occur less frequently than optic gliomas. Some brain tumors are never detected because they cause no symptoms. Others can induce seizures, changes in behavior, loss of sensation, visual disturbances, lethargy, or even coma. Another sign that a brain tumor may be present is recurrent headache, sometimes combined with vomiting. Headaches may occur for many reasons; not every headache is associated with a brain tumor. If a tumor is suspected because of clinical symptoms, a neurological exam helps to determine what type of functional impairment exists, which provides clues about the tumor’s location. Neuroimaging, usually an MRI or computed tomography (CT) scan, can confirm a diagnosis and provide more information about the nature and location of the tumor. Treatment varies depending on the type of tumor, its location, and the overall health of the individual. People with NF1 are at a somewhat increased risk of developing several types of uncommon tumors and malignancies. Because these tumors and cancers occur so infrequently, overall risk of a person with NF1 developing them remains low. The consensus is that routine presymptomatic screening for any of these tumors in people with NF1 is not necessary.1 Pheochromocytomas are extremely rare in the general population, but occur with greater-than-normal frequency in people with NF1. (Even so, they do not occur often enough to merit routine screening.) It is likely that pheochromocytomas develop when certain cells in the adrenal glands have mutations in both copies of the NF1 gene, causing complete loss of the protein neurofibromin.14,15 About 85 to 95% of pheocromocytomas develop in the inner portion of the gland, known as the adrenal medulla,6(p237) although they may occur in nerve tissue as well. The average age of diagnosis in someone with NF1 is 38 years.6(p238) If a pheochromocytoma is suspected, a doctor may first order blood and urine tests to measure levels of adrenaline and noradrenaline, and then order an imaging test (MRI or CT) to identify where the abnormality is located. Surgical removal of the tumor is usually the treatment of choice. If the tumor cannot be removed, a physician will prescribe medications to block the effects of the hormones and to control complications such as high blood pressure. Carcinoid tumors, like pheochromocytomas, secrete excess amounts of hormones. People with NF1 who develop pheochromocytomas are more likely to develop carcinoid tumors—and vice versa—although both types of tumors are uncommon in the NF1 population.16,17 If one type of tumor is diagnosed, a patient should be asked about symptoms that might indicate the other type has also developed. Carcinoid tumors are cancers that usually develop in the gastrointestinal tract, although they can occur elsewhere. These tumors secrete a variety of peptides that are involved in digestion. For the most part, these tumors grow slowly and do not cause symptoms for years, if ever. When symptoms do result, it is between the ages of 40 and 60 that they generally become evident.16,18,19 Black people with NF1 are more prone to have this tumor than are whites.20 Initial symptoms may include abdominal cramping and changes in bowel movements caused by obstruction of the intestine. If the tumor spreads to the liver, excessive skin flushing and dizziness may develop, probably because of excess release of hormones that dilate blood vessels. Treatment consists of surgical removal of the tumor and treatment with medications to control symptoms such as flushing. If the tumor has metastasized, it is incurable, but the cancer grows so slowly that individuals with this tumor may live for years. Juvenile myelomonocytic leukemia (JMML), formerly known as juvenile chronic myeloid leukemia, is a rare blood cancer of the hematopoietic cells, which are the precursors of red blood cells. JMML constitutes less than 1% of all childhood leukemias,21 but as many as 14% of these cancers22 occur in children with NF1. It is believed that JMML develops when NF1 gene function is lost, decreasing levels of neurofibromin, which both activates the Ras pathway (implicated in several cancers) and increases sensitivity to certain growth factors. This chain of events encourages myeloid precursor cells to proliferate out of control.23,24 As with other types of leukemia, treatment usually consists of chemotherapy. Bone marrow transplantation, though, may be necessary to ensure survival.25,26 Rhabdomyosarcomas are aggressive, malignant tumors that develop from myoblast cells, which are precursors of mature muscle cells. These tumors may occur throughout the body, although they most often develop in the head and neck, urinary and reproductive organs, limbs, and trunk. Most rhabdomyosarcomas develop in children and teenagers, with a peak incidence between 1 and 5 years of age.27 The tumor may first attract notice because it causes a visible lump. Those that are located deeper in the body cannot be seen, but they may have effects like eye protrusion, if located in the eye socket, or trouble urinating, if located on the bladder. It is not clear why rhabdomyosarcomas develop, but several lines of evidence suggest that loss or reduction of neurofibromin may prevent myoblasts from differentiating into mature muscle cells. Instead, they proliferate out of control.28–30 The usual treatment for rhabdomyosarcoma involves surgery, chemotherapy, and radiation. Chemotherapeutic agents include vincristine, cyclophosphamide, dactinomycin, Adriamycin, VP–16, and ifosfamide.27 For reasons that are unclear, people with NF1 may have a head that is larger in circumference than normal. Studies report that anywhere from 29 to 45% of people with NF1 have large heads, defined as greater than or equal to two standard deviations above the mean for their sex and age.2,31–33 Skulls are measured at the widest part, known medically as the fronto-occipital circumference. The head is usually enlarged relative to the rest of the body; someone of short stature, for instance, may have a head size in the “normal” range yet out of proportion to his body size. Only rarely is a large skull size related to an underlying disorder such as hydrocephalus (see below). The skull is larger than normal because the brain is also larger than average, although this seems to have no impact on cognition or other manifestations of NF1. The skull remains larger than average throughout the individual’s life. It is not known why the brain and skull grow larger than expected in a significant number of people with NF1. Head size has no correlation with the severity of NF1 symptoms, including cognitive impairment or learning disabilities. Some researchers have reported that children with NF1 have increased white matter (the part of the brain consisting of cells that support the gray matter, which consists of neurons).34 Autopsies have revealed that people with NF1 have more astrocytes, a type of support cell in the brain, than expected.35 Large head circumference should be noted if it occurs and monitored as a child grows older, but it is not cause for worry. The child’s skull usually grows at a rate that parallels a normal growth curve. Further evaluation and testing may be necessary if head growth should dramatically accelerate, or if the individual begins to experience headaches, demonstrate neurological abnormalities (such as disturbance of balance), or have papilledema (swelling of the optic nerve because of increased pressure inside the skull). In any of these situations, a thorough neurological exam and possibly an MRI scan can help to determine what may be causing such signs and symptoms. Headaches are common in about one in 10 people with NF1, although these are generally indistinguishable from those experienced in the general population.32,33 The exceptions to this general rule are headaches associated with some underlying CNS malady such as a brain tumor or a vascular anomaly. Tension headaches that come and go in episodic fashion are the most common type experienced, although people with NF1 may also suffer from migraine headaches. One study found that headaches in individuals with NF1 occurred more frequently between the ages of 7 and 14 and then became less common with age.36 If an individual with NF1 experiences multiple headaches, the best strategy for the patient (or parent) is to compile a “headache diary,” noting when headaches occur, how long they last, any notable symptoms, and any possible triggers. Note also whether there is a history of headaches among first-degree relatives (parents, siblings, and children). Migraine headaches may cause more than pain. Visual disturbances such as flashing lights, blind spots, or even visual hallucinations are not unusual. Many children with NF1 and migraines also experience stomachaches. If temporary neurological complaints such as limb weakness occur, note this as well. This information helps the physician to determine whether further testing is necessary. If the headaches are unusually severe or long-lasting, or causing significant problems, the physician may conduct a thorough physical examination, including a neurological and ophthalmological evaluation. Problematic symptoms include partial paralysis, impediment to reading, writing, speaking, or comprehension (collectively known as aphasia), and/or disturbances in perceptions. Depending on the results of the initial examinations, further testing, such as MRI of the brain or a cerebral angiogram, may be necessary. For the most part, however, headaches in people with NF1 are similar to and treated in the same way as headaches in the general population. Tension headaches may respond to over-the-counter pain medication. Management of migraine headaches may involve prevention with medications such as propranolol or amitriptyline and/or treatment of symptoms with medications such as sumatriptan. Reduction of light and stimulus may also help (see the discussion on pain management in the section Management in Chapter 12). Epilepsy is reported in anywhere from 3.5 to 7%2,32,33,37 of individuals with NF1, although in some cases it is related to NF1 and in other cases it is related to some other underlying condition. Epilepsy can cause seizures, convulsions, and sudden alteration of consciousness. It is not clear why epilepsy develops in people with NF1, and for the most part the type of seizures experienced are similar to those seen in the general population. Grand mal seizures, for instance, involve spasms of the trunk and extremities along with a loss of consciousness. Petit mal seizures are more subtle; the person may suddenly stop all activity and stare blankly into space. If epilepsy is suspected, a physician may order an MRI and an electroencephalogram (EEG), which measures the patterns of electrical activity in the brain. The physician may also order laboratory studies, such as blood and urine analysis, to rule out infections or irregularities with metabolism. In many cases, a first seizure is not treated; instead, the physician waits to see if another occurs. If a diagnosis of epilepsy is made, treatment generally consists of medications that prevent seizures. The abnormal buildup of fluid in the brain, known medically as hydrocephalus, is a possible complication of NF1. If too much fluid accumulates, it can compress areas of the brain, causing neurological difficulties. In people with NF1, hydrocephalus most often develops because of a condition known as aqueductal stenosis, a constriction of the small canal that connects the third and fourth ventricles, fluid-filled cavities within the brain. The constriction prevents cerebrospinal fluid from draining into the spinal cord as it normally would, and hydrocephalus may result. Aqueductal stenosis is revealed in ~1 to 2.5%2,32,33 of people with NF1, and usually develops between 6 and 35 years of age.6(p195) Although some people with hydrocephalus experience no symptoms, others may contend with headaches, vision changes, speech difficulties, disturbances in gait, and even seizures. Another sign that hydrocephalus may be present is a swelling of the optic nerve caused by intracranial pressure (papilledema), or an abnormal ophthalmological or neurological exam. To determine whether aqueductal stenosis is present, the physician orders an MRI of the brain. Treatment for hydrocephalus involves surgical insertion of a ventricular shunt to drain cerebrospinal fluid. Diane D.: “When Julie was 2 years old, she was diagnosed with aspiration, a condition where food goes into the lungs rather than to the stomach. No one could figure out why that was happening. We put her on a feeding tube. It still took a while for us to figure out why she was aspirating. “I kept a daily journal of her symptoms and it went along with her wherever she went—day care, school, and home. We made a simple chart of how much formula she consumed, whether she vomited, what other symptoms there were, and included a place for comments at the bottom. The symptoms varied. She was vomiting constantly. She sometimes vomited seven times a day. She also had problems talking, walking, and eating, with symptoms lasting for up to 2 weeks. When I reviewed the chart with our doctor in Boston, he thought that Julie might have a migraine syndrome. She was almost 4 then, but young enough that she couldn’t tell us if she was having headaches. There were all these other neurological signs. So the doctor prescribed propranolol, which helped. “We kept her on the feeding tube until she was 5, then weaned her off it and she started eating solid food. We went through a period of experimentation with the medications, but it was clear that she had to remain on the propranolol. We increased the dose as she got older. She’ll probably be on some type of medication for the migraines for the rest of her life. She’s still much more likely to complain about her stomach first rather than a headache. When she first woke up this morning, you could see by the look on her face that she did not feel well. She said, ‘Everything is blurry.’ Then she sat down to eat and said, ‘I can’t eat. I feel sick.’ People were surprised that Julie developed a migraine syndrome so young. She was just a year old. Usually this type of problem develops in puberty. “When Julie was 5, the doctors found an optic glioma. So far it has been asymptomatic, but they’re monitoring it. She has an MRI and undergoes visual field testing regularly. She also sees an NF specialist in Boston, a neurologist, and an ophthalmologist.”
Other Neural Tumors and Nervous System Abnormalities in Neurofibromatosis 1
♦ Optic Gliomas
Symptoms and Diagnosis
Decreased Visual Acuity
Decreased Color Vision
Afferent Pupillary Defect
Visual Field Defects
Papilledema
Optic Nerve Atrophy
Visual Evoked Responses
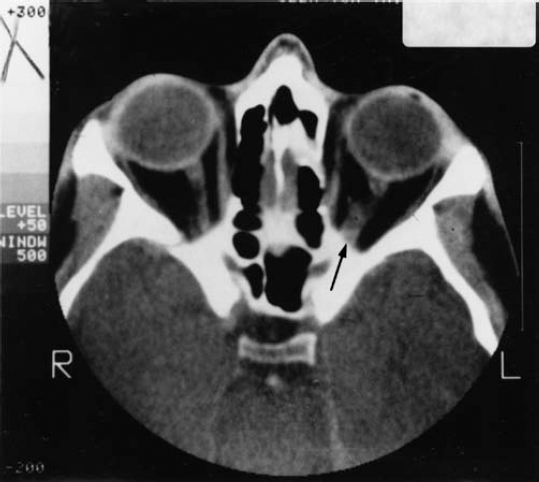
Management of Optic Pathway Gliomas
♦ Astrocytomas
♦ Other Malignancies in Neurofibromatosis 1
♦ Nervous System Abnormalities
Macrocephaly
Headaches
Epilepsy
Hydrocephalus
♦ The Personal Perspective
References
< div class='tao-gold-member'>
Other Neural Tumors and Nervous System Abnormalities in Neurofibromatosis 1
Only gold members can continue reading. Log In or Register to continue






Full access? Get Clinical Tree


