FIGURE 2.1 A. Gross brain. B. T-1 weighted MR image. Approximately matched midline sagittal sections. Sagittal T1-weighted MRI of the brain and upper cervical spinal cord. Labels: 1, septum pellucidum; 2, corpus callosum (a, genu; b, body; c, splenium; d, rostrum); 3, cingulate gyrus; 4, central sulcus; 5, column of fornix; 6, quadrigeminal plate; 7, quadrigeminal cistern; 8, thalamus; 9, cerebellum; 10, aqueduct of Sylvius; 11, fourth ventricle; 12, medulla; 13, pons; 14, mammillary body; 15, optic tract; 16, olfactory area; 17, gyrus rectus; 20, parietal-occipital fissure; 21, calcarine fissure; 49, midbrain; 51, pineal gland. (Reprinted with permission from Barboriak DP, Taveras JM. Normal cerebral anatomy with magnetic resonance imaging. In: Ferrucci JT, ed. Taveras and Ferrucci’s Radiology on CD-ROM. Philadelphia: Lippincott Williams & Wilkins, 2003.)
NEUROEMBRYOLOGY
In the embryo, development of the nervous system begins when ectodermal cells start to form the neural tube. The sonic hedgehog gene is vital for normal CNS development. It mediates a number of processes in development, including differentiation of the neuroectoderm. The neural tube begins to form in the 3rd week and is completed by the 4th week of embryonic life. The first stage in neural tube development is a thickening of ectoderm, forming the neural plate. A longitudinal fissure develops in the neural plate and progressively enlarges to form the neural groove. Differentiation of the cephalic from the caudal end of the neural groove is controlled by a signaling molecule called noggin. As the groove deepens, its edges become more prominent, and become the neural folds. The folds eventually meet, fuse, and complete the transformation into a tubular structure. The neural tube lies between the ectoderm on the surface and the notochord below. The cranial part of the neural tube evolves into the brain, and the caudal part becomes the spinal cord. With closure of the neural tube, the neural crest—neuroectoderm not incorporated into the neural tube—lies between the neural tube and the surface. Neural crest cells give rise to the PNS. Cells that lie ventral in the neural tube develop into motor cells, and those that lie dorsal develop into sensory cells. Sonic hedgehog is involved in this differentiation. Retinoic acid is also important at this stage, and the use of retinoic acid derivatives for acne treatment in early pregnancy may have catastrophic effects on the developing nervous system.
Neuroepithelial cells in the wall of the neural tube form neuroblasts, which develop into neurons, and glioblasts, which develop into macroglial and ependymal cells. With further maturation, the neural tube wall develops three layers: an innermost ventricular layer composed of ependymal cells, a mantle (intermediate) layer consisting of neurons and macroglia, and an outer marginal layer, which contains the nerve fibers of the neuroblasts in the mantle layer. The ventricular layer eventually forms the lining of the ventricles and central canal of the spinal cord, the mantle layer becomes the central gray matter, and the marginal layer becomes the white matter. Closure of the tube (neurulation) separates the developing nervous system from the surface ectoderm, forming the neurula (the embryo at 19 to 26 days after fertilization). Neurulation begins near the midpoint of the neural tube and advances toward the anterior (cephalic) and posterior (caudal) neuropores at either end; the anterior and posterior neuropores are the last sites to close. Neurulation is complete by 4 weeks; afterward, the CNS is a long, fluid-filled, tubular structure, and this basic configuration is maintained throughout life. Defective neurulation is common. Neural tube defects (NTDs) are common congenital malformations that result from failure of normal neural tube closure during early embryogenesis (Box 2.1). Neural tube closure is complete by the end of the 1st month; NTDs happen before a mother knows she is pregnant.
Neural Tube Defects
Neural tube defects (NTDs) are very common. They may be divided into an upper type (anencephaly, encephalocele) and a lower type (spinal dysraphism). Anencephaly is a lethal malformation that results from failure of closure of the anterior neuropore. The brain fails to develop. The face develops, but the cranial vault does not, and the brain may consist of only a tangled knot of primordial central nervous system tissue. Anencephaly is a common cause of stillbirth. There may be enough brainstem present to support vegetative life for a brief period. Failure of the posterior neuropore to close normally causes congenital malformations affecting the lumbosacral region. The most severe of these is myelomeningocele, essentially the posterior neuropore equivalent of anencephaly. The posterior elements of the lumbosacral vertebra fail to develop, the spinal canal is open posteriorly, and the spinal cord and cauda equina are herniated dorsally into a sac that lies over the surface of the lower back. The patients have severe neurologic deficits involving the lower extremities, bowel, and bladder. When the defect is less severe, the sac contains only meninges (meningocele). A mild defect of posterior neuropore closure results only in failure of normal fusion of the posterior arches of the lumbosacral vertebra. Patients are neurologically normal, and the defect is seen only on imaging studies (spina bifida occulta). Spina bifida occulta is quite common, affecting up to 10% of the population. Incomplete defects of anterior neuropore closure cause similar defects affecting the head and neck. An encephalocele is herniation of brain tissue through a bony defect in the skull. Encephaloceles most commonly occur in the occipitocervical region and are clinically obvious. When they involve the base of the skull (basal encephalocele) they may not be obvious. Cranium bifidum is dysraphism limited to the bony elements of the skull, most often the occipital bone; it is the cephalic analogue of spina bifida occulta. Arnold-Chiari malformations may involve defects in closure of both the anterior and posterior neuropore; these complex anomalies are discussed further in Chapter 21.
The pathogenesis of NTDs is multifactorial; both genetic and environmental factors are important, and the pattern of occurrence suggests a multifactorial polygenic or oligogenic etiology. Overactivation of sonic hedgehog signaling has been implicated. There are significant geographic differences, for example, NTDs are very common in Ireland. Folic acid plays a pivotal role in neuroembryogenesis. Genetic defects of the folate and homocysteine pathways have been implicated in the etiology of NTDs; periconceptional folate supplementation reduces the risk, and mothers of affected children may have elevated plasma homocysteine levels. A group at particular risk for having children with NTDs is women receiving certain antiepileptic medications during pregnancy.
The brain develops from the region of the anterior neuropore, forming three and then five vesicles. First, there is segmentation into three parts: forebrain (prosencephalon), midbrain (mesencephalon), and hindbrain (rhombencephalon) (Table 2.1). The forebrain then divides into the telencephalon, which becomes the cerebrum, and the diencephalon. The hindbrain divides into the metencephalon, which becomes the pons and cerebellum, and the myelencephalon, which becomes the medulla. The five-vesicle stage is complete by 6 weeks of embryonic life. The telencephalon then undergoes midline cleavage into a pair of side-by-side vesicles—primordial hemispheres. Regions of the telencephalon expand (evaginate) to form the cerebral hemispheres. The neural tube lumen continues into the evaginations, forming the ventricular system.
TABLE 2.1 The Derivatives of the Anterior Neuropore

Failure of normal cleavage into two hemispheres results in distinctive anomalies. Milder forms include arrhinencephaly, in which there is absence of the olfactory bulbs and tracts, and agenesis of the corpus callosum. Severe cleavage failure results in holoprosencephaly, in which there is only a single “hemisphere” (alobar prosencephaly), or a partial attempt at division (lobar and semilobar prosencephaly). Prenatal diagnosis is possible using sonography. The genes that control segmentation are also important in development of the face, and some anomalies involve both the face and the brain, particularly holoprosencephaly. Certain patterns of midline facial abnormality predict a severe brain malformation.
Following the segmentation and cleavage stages of neuroembryogenesis, the developing nervous system enters a stage of cellular proliferation and migration that is not complete until after birth. Neurons in the germinal matrix proliferate intensely, and then migrate to different parts of the nervous system. Cells destined to populate a specific brain region arise from a specific part of the germinal matrix. Processes that interfere with normal proliferation and migration cause another set of congenital malformations that includes microcephaly, megalencephaly, cortical heterotopia (band heterotopia, double cortex), agenesis of the corpus callosum, and schizencephaly. Three major callosal abnormalities have been identified: hypoplasia, hypoplasia with dysplasia, and complete agenesis. Finally, the brain develops its pattern of gyri and sulci. Defects at this stage of neocortical formation produce lissencephaly, in which the sulci and gyri fail to develop (smooth brain), pachygyria, in which the gyri are thicker than normal, and polymicrogyria, in which there are an excessive number of small gyri. These abnormalities may affect all or only part of the brain. Typically, children with these malformations have developmental delay and seizures. Other systems may be involved in these neuronal migration disorders, including eye and muscle (muscleeye-brain disease, Walker-Warburg syndrome, and Fukuyama congenital muscular dystrophy). Modern imaging, including prenatal magnetic resonance imaging (MRI), may identify some of these disorders.
Even after normal formation, the nervous system may be affected by intrauterine processes. In hydranencephaly, the hemispheres are destroyed and the remnants lie in a sac of meninges. This is to be distinguished from hydrocephalus, where the ventricles are markedly expanded. In hydranencephaly, the skull is normal but devoid of meaningful contents, in contrast to anencephaly, in which the skull is malformed along with the brain. In porencephaly, a cyst forms in a region where developing brain has been destroyed or has developed abnormally. Transillumination of the skull with a strong light may help detect these disorders early. The diagnosis may be confirmed by computed tomography, MRI, or sonography, and the diagnosis can be made with sonography in the prenatal period. Numerous conditions may affect the neonatal brain, including germinal matrix hemorrhage, hypoxic-ischemic encephalopathy, cerebral infarction, and infection. Many of these disorders produce “cerebral palsy,” an umbrella term with little neurologic meaning.
BONY ANATOMY
The skull is fashioned of several large bones and myriad complexly articulated smaller bones. The major bones are the frontal, temporal, parietal, occipital, and sphenoid; all are joined by suture lines. The major sutures are the sagittal and coronal, but there are numerous others. Sometimes sutures close prematurely (craniostenosis, craniosynostosis), before the skull has completed growth, producing malformed and misshapen skulls (Box 2.2; Figure 2.2). Noggin plays a role in the regulation of cranial suture fusion, and craniosynostosis may be the result of inappropriate down-regulation of noggin expression.
Craniosynostosis
The primary clinical manifestation of craniosynostosis is an abnormally shaped skull; the configuration depends on which suture(s) have fused prematurely. The skull is unable to expand in a direction perpendicular to the fused suture line. With synostosis of a major suture, the skull compensates by expanding in a direction perpendicular to the uninvolved sutures. Premature closure of the sagittal suture, the most common form of craniosynostosis, produces a skull that is abnormally elongated (scaphocephaly, dolichocephaly). Synostosis of both coronal sutures causes a skull that is abnormally wide (brachycephaly). When the coronal and lambdoid sutures are involved the skull is tall and narrow (turricephaly, tower skull). Synostosis of the sagittal and both coronal sutures causes oxycephaly (acrocephaly), a pointed, conical skull. Plagiocephaly refers to a flattened spot on one side of the head; it is due to premature unilateral fusion of one coronal or lambdoid suture. Synostosis involving the metopic suture causes trigonocephaly, a narrow, triangular forehead with lateral constriction of the temples. Synostosis of the posterior sagittal and both lambdoidal sutures produces the “Mercedes Benz pattern.” Severe craniosynostosis involving multiple sutures may cause increased intracranial pressure. Craniosynostosis usually occurs as an isolated condition, but there are numerous syndromes in which craniosynostosis occurs in conjunction with other anomalies, particularly malformations of the face and the digits, for example, Crouzon’s, Apert’s, and Carpenter’s syndromes. Several genetic mutations may cause craniosynostosis. There are many potential causes of nonsyndromic craniosynostosis, including environmental, hormonal, and biomechanical factors.
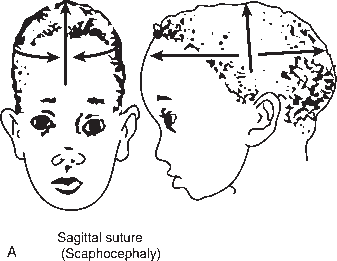
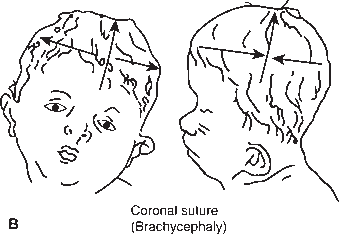
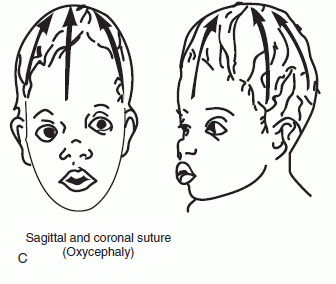
FIGURE 2.2 Craniosynostosis involving the cranial sutures: (A) sagittal; (B) coronal; and (C) both sagittal and coronal. (From http://www.artmouth.edu/∼dons/figures/chapt_1/Fig_1_2.htm)
The interior of the skull is divided into compartments, or fossae. The anterior fossa contains the frontal lobes, which rest on the orbital plates. The cribriform plate lies far anteriorly, between the orbital roofs; when fractured during head injury, cerebrospinal fluid (CSF) rhinorrhea may ensue. The middle fossa primarily contains the temporal lobes, and several major cranial nerves (CNs) run through the area. The posterior fossa contains the brainstem, cerebellum, and vertebrobasilar vessels. Except for CNs I and II, all the CNs run through or exit from the posterior fossa.
The frontal bone contains the frontal sinuses. The temporal bone has two parts: the thin squamous portion forms the temple; the thick petrous part forms the floor of the middle fossa. The squamous part contains the groove of the middle meningeal artery, and may be easily fractured, sometimes producing epidural hematoma. The petrous pyramids have their apices pointed medially and their thick bases pointed laterally; deep within are the middle and inner ear structures, the internal auditory meatus, the facial canal with its genu, and the air cells of the mastoid sinus. Fractures through the petrous bone may cause hemotympanum (blood in the middle ear cavity), hearing loss, or facial nerve palsy.
The sphenoid bone has greater and lesser wings and contains the sella turcica. The greater wings form the anterior wall of the middle fossa; the lesser wings form part of the floor of the anterior fossa. The greater and lesser wings attach to the body of the sphenoid, buried within which is the sphenoid sinus cavity. The best way to appreciate the anatomy of the sphenoid bone is to look at it in disarticulated isolation, when the “wings” become obvious. The sella turcica makes up a saddle-shaped depression in the body of the sphenoid; alongside the sella lie the cavernous sinuses. The pituitary gland lies within the sella, and neoplasms of the pituitary may enlarge the sella and push upwards out of the sella onto the optic chiasm. Enlargement of the sella is a nonspecific finding in increased intracranial pressure.
The occipital bone makes up the posterior fossa. The clivus forms the anterior wall of the posterior fossa; it ends superiorly in the dorsum sellae and posterior clinoid processes. The basilar artery and brainstem lie along the clivus. Tumors, most often chordomas, may erode the clivus and produce multiple CN palsies. Various structures pass into or out of the skull through the numerous foramina that pierce its base (Table 2.2). Pathologic processes may involve different foramina; the resultant combination of CN abnormalities permits localization (see Chapter 21).
TABLE 2.2 Major Skull Base Foramina and Their Contents
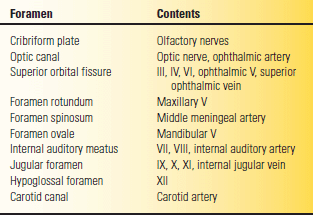
Roman numerals refer to cranial nerves III through XII.








