CHAPTER 71 PARKINSON’S DISEASE
Parkinson’s disease (PD) is a neurodegenerative disease with initial clinical features that are predominantly the result of loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) of the midbrain. As the disease progresses, the involvement of additional brain areas in the degenerative process produces mainly nondopaminergic, nonmotor features. The discovery of dopamine deficiency in PD and the introduction of levodopa have provided patients with a significant improvement in both quality of life and life expectancy, but the treatment of nonmotor features and slowing of disease progression remain important unmet needs for patients.
HISTORICAL PERSPECTIVE
In 1960, Ehringer and Hornykiewicz identified the dopamine deficiency in PD striatum.1 Studies on the replacement of dopamine with DL-dopa produced equivocal results until used in sufficient quantity.2 This began the era of symptomatic treatment for PD, which has remained focused on the dopaminergic system for almost 40 years.
EPIDEMIOLOGY
Defining the epidemiology of PD is confounded by several variables that include the difficulty in diagnosis and the age dependence of the disease. Several studies have sought to define incidence. In the United States, the age-adjusted figure is 13.5 to 13.9 per 100,000 person-years.3,4 The age-adjusted prevalence is approximately 115 per 100,000 and is estimated as 1.3 per 100,000 under age 45 years and 1192.9 per 100,000 in those aged 75 to 85 years.3 A prevalence study in Holland found 3100 cases per 100,000 aged 75 to 85 years and 4300 per 100,000 for those over age 85 years.6 The geographical distribution of the disease appears similar across the United States and Japan, but failure to adjust population figures for age can lead to widely discrepant results, such as the prevalence of 10 per 100,000 in Nigeria.7
PATHOLOGY
Macroscopic examination of the sectioned PD brain shows depigmentation of the substantia nigra and locus ceruleus. The characteristic pathological change of PD is the loss of pigmented dopaminergic neurons, particularly in the ventral tier of the SNc with intracytoplasmic eosinophilic inclusions (Lewy bodies) in a proportion of the surviving neurons (Fig. 71-1). The SNc also contains activated microglia and extracellular neuromelanin. There is also cell loss in the locus ceruleus; the nucleus basalis of Meynert; the dorsal motor nucleus of the vagus; the Edinger-Westphal, raphe, and pedunculopontine nuclei; and the pons, midbrain, spinal cord, and peripheral sympathetic ganglia. Lewy bodies may be found in these areas and in the cortex. Thus, in addition to the dopaminergic system, the cholinergic, serotonergic, γ-amino butyric acid, and adrenergic transmitter systems are involved in PD.
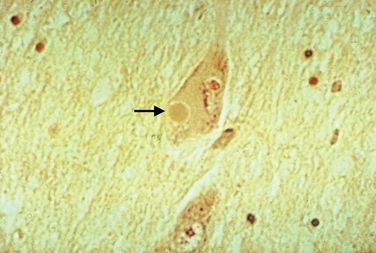
Lewy bodies have attracted considerable attention over the years, as they may hold important clues to pathogenesis of the disease. They are 5 to 30 μm in diameter with a hyaline eosinophilic core, which may be dense and composed of concentric lamellae; a pale halo may be seen around the core. Electron microscopy demonstrates 7-to 20-nm intermediate filaments. The Lewy body is composed of a number of different proteins, staining for ubiquitin, α-synuclein, and proteasomal components. It is not known whether these inclusions represent a protective response to aggregate abnormal or toxic proteins, or whether their formation is part of a toxic process that damages the cell.
Studies suggest that the earliest pathological changes are seen in the dorsal motor nucleus and in the olfactory bulbs and nucleus—Braak stages 1 and 28 (Fig. 71-2). In this context, it is noteworthy that loss of olfactory function can occur at a time prior to the onset of dopaminergic symptoms or signs and may serve to define an “at-risk” population.9 Lewy bodies then develop in the locus ceruleus and progress in the medulla and pons.10 The appearance of inclusions in the SNc defines the onset of Braak stage 3 with progression to stage 4. At this stage there is also degeneration in the pedunculopontine nucleus, the dorsal raphe nuclei, and the hypothalamus. Stages 5 and 6 involve progressive involvement of the cerebral cortex and neurodegeneration in those regions already affected.
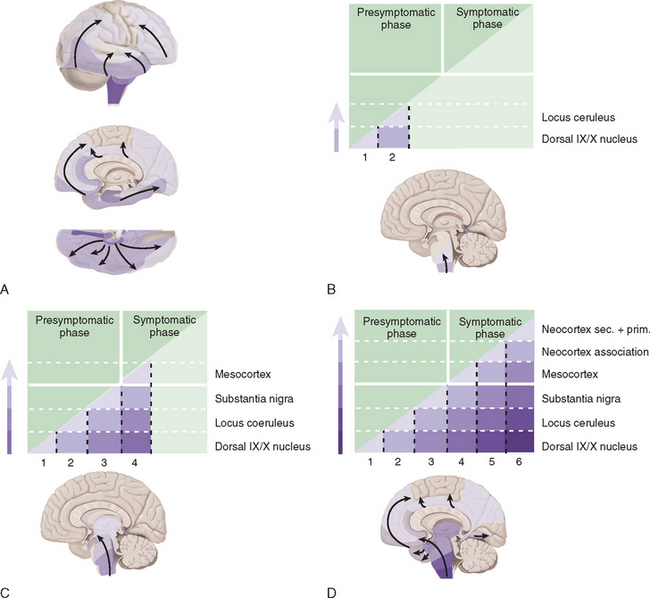
Figure 71-2 Braak staging of Parkinson’s disease.
(From Del Tredici K, Rub U, De Vos RA, et al: Where does Parkinson’s disease pathology begin in the brain? J Neuropathol Exp Neurol 2002; 61:413-426; and Braak H, Del Tredici K, Rub U, et al: Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24:197-211.)
ETIOLOGY
Genetic Factors
Sir William Gowers recognized that there was an increased prevalence of PD among the relatives of sufferers and proposed a genetic cause.11 Epidemiological studies evaluating the genetic contribution of PD are complicated by ensuring an accurate clinical diagnosis, inability to identify presymptomatic cases and the need for an appropriate control population. Case-control studies have confirmed Gowers’ observation that PD is more common in relatives of PD cases compared with matched controls.12–16 The relative risk of developing PD in family members has varied widely between different studies, but in many, incomplete family information was obtained and only one study has been population based.16 Overall, the relative risk in first-degree relatives of PD cases is increased approximately two- to threefold.17
A large PD twin study showed no significant concordance for PD among monozygotic twins, suggesting that there was no significant genetic contribution to PD.18 However, there was significant concordance for those with onset before age 50 years, implying that young-onset PD is more likely genetically determined. Another smaller twin study using fluorodopa positron emission tomography (PET) to image dopaminergic function in both affected and unaffected monozygotic and dizygotic twin pairs demonstrated an increased concordance for PD among identical twins.19 At follow-up, the combined concordance levels for subclinical dopaminergic dysfunction and clinical PD were 75% in the 12 monozygotic twins and 22% in the 9 dizygotic twins evaluated twice.
Table 71-1 shows the current list of genes known to cause PD. PARK1 through PARK9 have been discovered using large single or combined pedigrees, whereas PARK10 and PARK11 have been identified using association techniques.
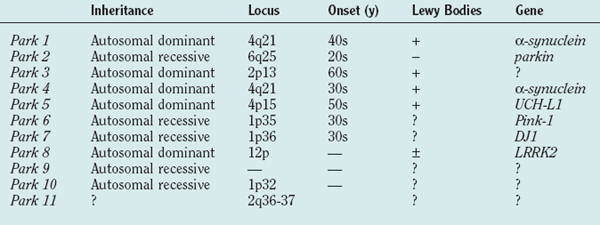
α-Synuclein (PARK1)
PARK1 involves mutations in the α-synuclein gene.20,21 The first families with an α-synuclein mutation originated from southern Italy and Greece, and with the description of several additional families with the same A53T mutation, it is believed that they have a common founder. Age at onset was young (mean, 46 years) with a low frequency of tremor and relatively rapid disease progression, although symptoms were levodopa responsive.22 Pathological analysis of the few brains available revealed the presence of Lewy bodies in the substantia nigra and locus ceruleus.23 Linkage to markers on chromosome 4q21-q23 was demonstrated and the locus designated PARK1.24 Further analyses identified a mutation in exon 4 of the gene encoding α-synuclein resulting in an alanine-to-threonine substitution at codon 53 (A53T), which was also found in affected members of three Greek families with earlyonset autosomal dominant PD.20 A second mutation (A30P) was later found in a small German family with PD,21 but extensive study in large groups of PD families and sporadic cases has not identified other patients or families with this mutation.25–28
α-Synuclein is a protein of 140 amino acids that is predominantly expressed in neurons and is one of the most common brain proteins. Its normal function remains unclear, although it plays a role in synaptic plasticity based on song-learning studies in the zebra finch29 and vesicular regulation of dopamine release from knockout mice.30 A key observation linking α-synuclein to PD was the demonstration that it is one of the principal components of Lewy bodies.31 Lewy bodies are intracytoplasmic aggregates comprising several proteins, including ubiquitin and α-synuclein, and have supported the notion that abnormal protein handling might be important in PD pathogenesis. Furthermore, mutant isoforms of α-synuclein more readily oligomerize and it has been suggested that its tendency to aggregate into misfolded structures may confer toxic properties to the protein. Indeed, overexpression of wild-type or mutant protein in transgenic mice or Drosophila reproduces many of the behavioral and pathological features of PD.32,33
Multiplications of the wild-type α-synuclein gene have been described in PD families. A triplication of the gene was identified in a large autosomal dominant kindred with PD and tremor,34 and duplication of the gene was found in one of 42 familial probands of early onset PD.35 A third α-synuclein point mutation (E46K) has been reported in an autosomal dominant family with parkinsonism and Lewy body dementia.36
Several models of abnormal α-synuclein expression have been developed. Knockout of the gene in mice resulted in no detectable abnormality other than an alteration of dopamine release in response to rapid stimulation, although this has no clear functional correlate.30 Overexpression of wild-type human α-synuclein in mice resulted in loss of dopaminergic terminals, intranuclear and cytoplasmic ubiquitin-rich nonfibrillar α-synuclein inclusions in the substantia nigra, hippocampus, cortex, and a rotor-rod motor deficit at 1 year.32 Overexpression of human wild-type and mutant α-synuclein in flies caused a loss of dopaminergic neurons, Lewy body–like inclusions with fibrillar α-synuclein, and a motor deficit with no significant difference between wild-type and mutant α-synuclein.33 Additional mouse models of α-synuclein expression have demonstrated inclusion formation and spinal cord pathology but no dopaminergic cell loss, and motor deficit at late stage.37 Virus-mediated overexpression of α-synuclein induces nigral degeneration in rodents.38
The α-synuclein mutations result in the protofibrillar form, which is considered the more toxic form of the protein. The A53T and A30P α-synuclein mutations promote protofibril formation and A30P inhibits conversion to fibrils.39 Catecholamines, including dopamine and levodopa, inhibit fibril formation in vitro, and this is reversed by antioxidants; that is, catechol oxidation promotes protofibril formation.40 This observation would support a protective role for Lewy bodies in PD. An important observation revealing a potential toxic mechanism for α-synuclein is that the mutant A30P form increases toxicity to dopamine, increasing cell death and free radical–mediated damage.41 The authors proposed that the mutation impaired vesicular uptake of dopamine, resulting in higher cytoplasmic or extravesicular synaptic concentrations of dopamine that would in turn cause free radical–mediated damage. Phosphorylation at the Ser129 residue is required to mediate the toxicity of α-synuclein and increased the formation of inclusions in SHSY-5Y cells.42 This phosphorylated form of α-synuclein is present in Lewy bodies.43 Prevention of this phosphorylation by substitution of an alanine residue reduced inclusion formation in the SHSY-5Y model, and in the Drosophila model this same mutation at 129 that prevents phosphorylation protected against dopaminergic neuronal loss.44
Parkin (PARK2)
PARK2 gene mutations were first identified in autosomal recessive juvenile-onset parkinsonism (ARJPD).45 ARJPD has been most commonly seen in the Japanese population and is characterized by onset before age 40 years, symptomatic improvement following sleep, mild dystonia, and a good response to levodopa.46 Resting tremor is seen less frequently than in idiopathic PD, and patients may have brisk tendon reflexes but no other pyramidal, cerebellar, or autonomic features. The disease is often symmetrical and dyskinesias develop early but progression is usually slow. Pathologically, there is dopaminergic cell loss in the SNc and locus ceruleus but no Lewy bodies are seen.47 The gene responsible for ARJPD was mapped to 6q25.2-q27,48 and in 1998 the gene was discovered and named parkin.45 Affected patients carry deletions or point mutations in various parts of the parkin gene.49,50 The absence of Lewy bodies in ARJPD may simply reflect the limited time over which the pathology has evolved. However, the relationship of parkin mutations to idiopathic PD has been highlighted by the identification of parkin mutations in apparently sporadic cases of PD and by the description of Lewy bodies in parkin positive patients with later-onset disease than ARJPD.51,52 Parkin mutations are a common cause of PD under age 25 years but rare over age 40 years.53,54 Parkin-related PD has been reported in multiple generations in families without consanguinity, suggesting a pseudo-autosomal dominant mode of inheritance for some mutations.55,56 Fluorodopa positron emission tomography (PET) in parkin patients demonstrates reduced uptake in the striatum, although there is some discordance regarding the symmetry and pattern of this reduction57,58 However, the rate of loss of fluorodopa PET signal was slower in the parkin patients than in sporadic PD.59 Asymptomatic heterozygous parkin mutation carriers had intermediate levels of striatal fluorodopa PET uptake compared with normal controls and homozygous symptomatic patients.58 This suggests an intermediate stage of nigrostriatal dysfunction that may interact with other genetic or environmental factors to induce PD. The frequency of heterozygous parkin mutation carriers is not known.
PARK2 encodes parkin, which functions as an E3 ligase, ubiquinating proteins for destruction by the proteosome.60,61 Several substrates for parkin have been identified, including a 22-kDa glycosylated form of α-synuclein, parkin-associated endothelin receptor-like receptor (Pael-R), and CDCrel-1. Overexpression of Pael-R causes it to become ubiquinated, insoluble, and unfolded and leads to endoplasmic reticulum stress and cell death.62 It has been demonstrated to accumulate in its insoluble form in the brains of patients with parkin mutations, suggesting a possible toxic mechanism. CDCrel-1 is a protein involved in cytokinesis and may influence synaptic vesicle function.61 Overexpression of parkin protected against dopaminergic loss in rodents coexpressing α-synuclein, suggesting a protective role for parkin.63
A parkin knockout mouse model has been described.64 This showed an increase in striatal extracellular dopamine, a reduction in synaptic excitability, and a mild nonprogressive motor deficit at 2 to 4 months. There was no loss of dopaminergic neurons and no inclusion formation. Dopamine receptor binding affinities and parkin E3 ligase substrate levels were normal. Interestingly, these mice had decreased striatal mitochondrial respiratory chain function and reductions in specific respiratory chain and antioxidant proteins.65 Parkin knockout flies developed muscle pathology, mitochondrial abnormalities and apoptotic cell death.66 Overexpression of parkin in PC12 cells indicated that it is associated with the mitochondrial outer membrane.67 Parkin-positive patients have decreased lymphocyte complex I activity.68 The ability of parkin to ubiquinate proteins may be impaired by S-nitrosylation, which in turn may be a consequence of excitotoxicity-mediated damage.69
UCH-L1 (PARK5)
A further mutation in the gene encoding ubiquitin carboxyhydrolase (UCH)-L1 again supported the relevance of the ubiquitin-proteosomal system (UPS) in PD pathogenesis.70 UCH-LI is an enzyme that hydrolyzes the C-terminus of ubiquitin to generate ubiquitin monomers that can be recycled to clear other proteins. A missense mutation was identified in two siblings with typical PD in a German family demonstrating apparent autosomal dominant inheritance.70 Age at onset was 49 years in one and 51 years in the other. The mutant form of UCH-L1 was shown to have reduced enzyme activity resulting in impaired protein clearance through the ubiquitin-proteasome pathway. However, no other mutations in this gene have been identified in other families, suggesting it is a rare cause of PD.71,72 Given that no further cases of PD have been described with mutations in this gene, some doubt has been cast on the relevance of UCH-L1 to PD.
PINK1 (PARK6)
The PARK6 locus (chromosome 1p3673) was first identified in a large consanguineous Italian family and subsequently in an additional three Italian families and others from Europe and elsewhere, including Asia.74–78 The mean age at onset ranges from 21 to 57 years. Progression is usually slow and patients exhibit a good response to levodopa. PARK6 mutations appear to be a rare cause of PD.
The PINK1 (PTEN-induced kinase 1) gene is ubiquitously transcribed and is believed to encode a mitochondrial kinase.74,79 It is believed that PINK1 may play a role in protecting cells against stress conditions that affect mitochondrial membrane potential, but the downstream targets through which PINK1 mediates its protection have not been identified. As 11 of the 14 reported mutations fall into the kinase domain of PINK1,74,76,77 altered phosphorylation of target proteins probably represents a key pathogenic mechanism, leading to abnormal stress response and neurodegeneration. The reversible phosphorylation of proteins is an important method of regulating cellular activities.80 Up to 30% of eukaryotic proteins are phosphorylated,81 and there are more than 500 human genes encoding protein kinases.82 The phosphorylation of mitochondrial proteins is considered pivotal to the regulation of respiratory activity in the cell and to signaling pathways leading to apoptosis, as well as for other vital mitochondrial processes. For instance the phosphorylation of α-synuclein is an important step in mediating its toxicity (see earlier), and Lewy bodies do contain the phosphorylated form of this protein.
DJ-1 (PARK7)
The PARK7 locus on chromosome 1p36, only about 25 cM from the PARK6 locus, was first identified in a small group of young-onset PD patients in a remote region of Holland.83 Average age at onset is 32 years, with a currently reported range of 25 to 40 years. Onset is asymmetrical, progress is slow, and there is a good response to levodopa. Tremor is infrequent, and psychiatric disturbances have been described in some. Fluorodopa PET scans demonstrate a symmetrical reduction in uptake. No pathological studies of PARK7 patients have been undertaken at the time of writing.
PARK7 encodes DJ-1; mutations are autosomal recessive and comprise both deletions and point mutations that result in a loss or inactivation of the protein. Its function is unknown, but it is widely distributed and conserved. It can protect against toxicity mediated by free radicals and transfers to the outer mitochondrial membrane under conditions of oxidative stress.84,85 Wild-type DJ-1 is also located in the mitochondrial matrix and intermembraneous space, and this distribution is not altered by mutations in the protein.86
LRRK2 (PARK8)
Mutations in the LRRK2 gene are the most common cause of either familial or “sporadic” PD identified to date. The LRRK2 G2019S mutation alone has been reported in 2.8% to 6.6% of autosomal dominant PD families87–89 and in 2% to 8% of sporadic cases.90–92 The G2019S mutation has variable penetrance, with 17% at 50 years and 85% at 70 years, a profile that mimics idiopathic, sporadic PD. Although other LRRK2 mutations are described, the G2019S mutation remains the most common cause of either sporadic or familial PD. This mutation has not been seen in Alzheimer’s disease or in parkinsonian syndromes other than idiopathic PD.93,94
Many of the reported cases of LRRK2 mutations have typical features of PD with asymmetrical onset of tremor, bradykinesia, and rigidity. As noted, the age at onset is variable with occasional very late onset cases89 and a report of one carrier male reaching 89 years with only subtle neurological changes.95 Patients have a good response to levodopa but develop motor complications including dyskinesias. Fluorodopa PET and imaging using ligands for the dopamine transporter with single-photon emission computed tomography (SPECT) demonstrate changes typical of those seen in idiopathic PD.96 Although all LRRK2 mutant brains examined to date demonstrate loss of dopaminergic neurons in the SNc, one of the morphological hallmarks of idiopathic PD, additional pathology may also be seen. Pure nigral neuronal degeneration was found in the first family linked to this locus97; neurofibrillary tangles, abnormal tau deposits, and widespread Lewy body synucleinopathy have been described in others, including one family with anterior horn cell loss.98,99 Three brains of “sporadic” PD with G2019S LRRK2 mutations have had pathological examination and all have demonstrated nigral neuronal loss and Lewy body formation typical of PD.90 All of these subjects had PD based on clinical criteria.
The LRRK2 gene encodes a 286-kDa cytoplasmic protein that is widely expressed in the brain.100 LRRK2 is a member of the ROCO protein family and appears to have multiple functions, at least by virtue of its predicted structure. These include a Ras/GTPase domain involved in cytoskeletal responses to external stimuli, vesicular trafficking and the stimulation of stress-activated kinase.101 The leucine-rich motif may have numerous functions, including protein-protein interactions and substrate binding for ubiquitination. The LRRK2 kinase domain belongs to the MAPKKK family of kinases with catalytic activity for both serine/threonine and tyrosine residues.
The G2019S mutation changes a highly conserved glycine at the start of the kinase activation segment, and it has been postulated that this has an activating effect causing a “gain of function” compatible with its autosomal dominant inheritance pattern.89 LRRK2 also has a WD40 domain, which again may be involved in cytoskeletal assembly and signal transduction.
PARK9, PARK10, and PARK11
The PARK9 locus on chromosome 1p36 was described in an autosomal recessive, juvenile-onset parkinsonian disorder with pyramidal features, ophthalmoplegia, and dementia.102,103 PARK10 on chromosome 1p32 was identified in the families of Icelandic PD patients with late-onset disease.104 PARK11 was obtained by association studies, and little information is available on phenotype.
Genetic Associations
Only a minority of cases of PD are part of a clear familial pedigree. Some of the single-gene mutations described above may account for a proportion of the remaining patients. However, our current understanding is that such single-gene causes of PD will remain in the minority. Thus, the large proportion of PD patients may develop their disease as a result of environmental factors, polygenic influences, or a combination of the two. There have been several genetic association studies attempting to determine significant polymorphisms that may increase or decrease the risk for PD. Further evidence for the role of genes in PD comes from genome-wide screens.105,106 The first found 174 families with a minimum of two clinically affected individuals with PD per family and identified a marker from the intronic region of parkin in an earlyonset group and a region on 9q in a dopa-resistant group. In the total sample, areas of interest were found on chromosomes 5q, 8p, and 17q. Data from the second genome-wide linkage study used a sample of 113 affected sibling pairs with PD and identified suggestive linkage on chromosomes 1, 9, 10, and 16, with no evidence implicating the regions containing parkin, α-synuclein, or tau genes. However, additional studies have shown that α-synuclein promoter region variants can influence the risk for PD.107,108 Those alleles that increase α-synuclein expression lead to an increased risk for developing PD, an observation in line with the multiplications of the gene causing familial PD (see earlier). Similarly, variants that influence parkin expression can also modulate the risk for PD: in this case, those alleles that lower parkin expression enhance the risk for PD.109 Mutations in the gene for glucocerebrosidase have been associated with an increased risk for PD among Ashkenazi Jews.110 Glucocerebrosidase deficiency causes type 1 Gaucher’s disease. Thirty-one of 99 Ashkenazi PD patients had one or two mutant alleles of the glucocerebrosidase gene (GBA), and 95 of 1543 controls (Ashkenazi) were carriers. Those with GBA mutations had onset of their PD around age 60 years and clinical features typical of idiopathic disease.
Genetic Causes of Parkinsonism
Several disorders that include parkinsonism in their phenotype have been characterized at the genetic level (Table 71-2). The frontotemporal dementias are discussed in detail in Chapter 73, the dystonias in Chapter 35, Huntington’s disease in Chapter 67, Wilson’s disease in Chapter 108 and the inherited ataxias in Chapter 68, so these will be discussed only briefly here.
TABLE 71-2 Secondary Familial Parkinsonism
The frontotemporal dementias and parkinsonism linked to chromosome 17 (FTDP-17) usually have onset in the fifth decade, although the age range is wide (25 to 75 years). Symptoms are of gradual onset and include motor dysfunction in the forms of parkinsonism, behavioral, and personality disorders and cognitive decline.111 Patients may exhibit apathy, depression, aggression, disinhibition, obsessive-compulsive disorder, executive dysfunction, and nonfluent aphasia. Patients and families tend to fall into either the predominantly parkinsonian or dementia types. Pathological examination shows severe frontotemporal atrophy and degeneration that includes the substantia nigra and the basal ganglia. There are tau accumulations in the remaining neurons and glia. FTDP-17 is autosomal dominant and is due to mutations in the tau gene. Although the genotype-phenotype relationship is relatively loose, those with the parkinsonism predominant form more commonly have exon 10′ or 5′ mutations.
Certain of the spinocerebellar ataxias (SCAs) are associated with parkinsonism and indeed may even manifest with this feature. SCA2 dopa-responsive parkinsonism is most often observed in the Chinese Asian population.112,113 Patients may have asymmetrical disease; a resting tremor and the presence of ataxia and abnormal eye movements may make differentiation from other parkinsonian disorders difficult if genetic testing is not performed. Imaging with fluorodopa PET has produced variable results from changes typical of those seen in PD114 to severe involvement of the caudate.115 The demonstration of an abnormally expanded CAG repeat in the ataxin-2 gene confirms SCA2. SCA3 (Machado-Joseph disease) mutations in conjunction with parkinsonism have been found most often in Caribbean populations.
Fragile X mental retardation complex is a common cause of mental retardation. It is an X-linked disease caused by an abnormal CGG expansion in the FMRI gene, which results in reduced gene expression. Intermediate length repeats can be a cause of tremor-ataxia parkinsonism in men. About 60% of these patients have a postural tremor, ataxia, autonomic dysfunction, impaired cognition, and symmetrical parkinsonism.116,117
Dystonia in association with parkinsonism is seen in a number of genetic diseases. X-linked dystonia parkinsonism was first reported in men from an island in the Philippines who had early onset of action tremor, dystonia, blepharospasm, and parkinsonism in 40% (Lubag) with poor response to levodopa. This disease is referred to as DYT3.118 Rapid-onset dystonia parkinsonism (DYT12) is an autosomal dominant disorder associated with bulbar features including dysarthria and dysphagia, dystonia, postural instability, and bradykinesia. Symptoms progress rapidly over hours and may be precipitated by physical or emotional stress. There is usually a poor response to levodopa. Mutations in the ATP1A3 gene that encodes a subunit of the sodium-potassium channel have been described in this disorder.119
Mitochondria have their own DNA, and the mitochondrial genome encodes 13 proteins of the oxidative phosphorylation system in addition to 2 ribosomal and 22 transfer RNAs. The discovery of complex I deficiency in PD substantia nigra (see later) raised the possibility that the mutation of genes (nuclear or mitochondrial) encoding complex I subunits might be involved in determining the enzyme’s defective activity. As mitochondrial DNA (mtDNA) is inherited in a strictly maternal pattern, if there were full penetrance of such a mtDNA gene defect, mitochondrial inheritance should be identifiable in pedigrees with parkinsonism. Such maternal inheritance has been described in PD120 but appears rare. However, it is known that 40% of patients with proven mitochondrial diseases and mtDNA mutations present as sporadic cases. Thus maternal inheritance is not a sine qua non of mtDNA gene defects. However, molecular genetic investigations of mtDNA have so far been unable to identify any specific mutation that clearly co-segregates with PD.
Studies using age-matched controls found no increase in the 5-kilobase mitochondrial deletion mutation in PD substantia nigra.121 Several studies have sequenced mtDNA in PD, but these have all used unselected patients in terms of their complex I activity.122,123 Although some reports have suggested an increased frequency of certain mtDNA polymorphisms in PD, this has not been replicated in all studies.124–128 Two studies have demonstrated a relationship between mtDNA haplotypes and the risk for developing PD. The first showed a reduced risk for PD in individuals with haplotypes J and K,129 and the second, a 22% decrease in PD in those with the UKJT haplotype cluster.130 In contrast, a smaller study reported an increased risk for PD with haplotypes J and T.131
Mutations in the gene for mtDNA polymerase gamma (POLG) have been demonstrated in patients with progressive external ophthalmoplegia and parkinsonism. Autosomal dominant or recessive inheritance of progressive external ophthalmoplegia with age at onset ranging from 10 to 54 years was followed some years later (range, 6 to 40 years) by the development of an asymmetrical, levodopa-responsive bradykinetic rigid syndrome together with resting tremor in some patients. Additional features included variable limb, pharyngeal or facial weakness, cataracts, ataxia, peripheral neuropathy, and premature ovarian failure.132 Muscle biopsy demonstrated ragged red, cytochrome oxidase–negative fibers in all patients with multiple mtDNA deletions on Southern blotting. Symmetrically reduced striatal 18-fluorodopa PET was seen in two patients. Brain histology was available on an additional two patients; both showed severe loss of substantia nigral dopaminergic neurons but without the development of Lewy bodies or other synuclein aggregates. Four families had the same A2864G mutation inherited in autosomal fashion in three and with a founder effect in the fourth. Mutations in the exonuclease or polymerase portions of the gene were identified in the autosomal recessive families. Another patient with autosomal dominant progressive external ophthalmoplegia parkinsonism and an A2492G mutation has been reported.133
Environmental Factors
Several studies have sought to define the environmental contributions to the etiology of PD. A rural residency appears to increase the risk of the development of PD and, in particular, young-onset PD.134–136 However, this finding has not been confirmed in all studies.137 Rural living is associated with farming and pesticide use, and an association with the agricultural industry has been found with increased incidence in PD patients.138–140 In addition, another lifestyle study showed increased herbicide exposure in patients with PD.141 Organochloride pesticides were identified as risk factors in a German case-control study142 with the offending agent being identified as the organochloride dieldrin, which was found in 6 of 20 PD brains and none of 14 control brains.143 Another study identified dithiocarbamates as a risk factor for PD,137 a compound that has also been shown to enhance 1-methyl 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity.144 Some studies have found that the significant association of PD with farming as an occupation cannot be accounted for by pesticide exposure alone.139 Another rural factor that has been linked to PD is the consumption of well water,145 although this may simply be further evidence in support of herbicides or pesticides as etiological factors for PD.
Carbon monoxide is a common environmental pollutant that may also play an important role in cell signaling. Acute carbon monoxide poisoning results in the complexing of carbon monoxide with the ferrous iron, protophorphyrin IX, and this prevents the carriage of oxygen. In addition, carbon monoxide is a potent inhibitor of cytochrome oxidase (complex IV) of the mitochondrial respiratory chain. Survivors of carbon monoxide poisoning have developed parkinsonism within a few days or weeks of exposure.146 The affected patients show necrosis of the globus pallidus on computed tomography scanning and magnetic resonance scanning.
Pyrethroid pesticides, when administered parenterally to rodents, cause a reduction in tyrosine hydroxylase–positive dopaminergic neurons in the nigrostriatum and an increase in dopamine transporter and brain-derived neurotrophic factor expression.147–150 These results indicate that commonly used pesticides can cross the blood-brain barrier and induce damage to the basal ganglia. Rotenone, a pesticide commonly used in the United States, when infused into rodents, can result in degeneration of nigrostriatal neurons and the formation of α-synuclein–rich Lewy-like inclusions.151 Paraquat, a widely used herbicide, has been shown to increase α-synuclein fibril formation in vitro in a dose-dependent fashion and to increase α-synuclein protein expression in mice with the reversible development of aggregates in substantia nigra dopaminergic neurons.152 Anonnacin, a component of sour-sop in the Caribbean, has been shown to produce a PD-like phenotype in humans and nigrostriatal loss in animals.153 MPTP, a meperidine analog designer drug, is known to produce parkinsonism in humans, other primates, and rodents through uptake and conversion mechanisms that target the nigrostriatal pathway,154 It is noteworthy that these agents result in inhibition of mitochondrial NADH CoQ reductase (complex 1) and are free radical generators, features of direct relevance to idiopathic PD.
Manganese is a constituent of several pesticides and herbicides as well as being an anti-knock additive to lead-free petrol. Manganese is neurotoxic to the basal ganglia and produces a parkinsonian syndrome with damage predominantly to the globus pallidum. There have been numerous reports of manganism developing among individuals exposed to manganese dioxide ore, usually by inhalation of manganese dust. Exposure is typically chronic over 6 months to 16 years, and the onset of manganism is slow, beginning with apathy, muscle weakness and cramps, and general irritability. Progression occurs and is characterized by dysarthria and psychosis followed by severe rigidity, anarthria, and dystonia.155 Manganese predominantly appears to affect the globus pallidus with sparing of the substantia nigra.156 There has been interest in the potential for manganese containing steel to induce parkinsonian features in welders,157 although this association is controversial. At present, it is not known if manganese-containing pesticides can induce PD.
Between 1917 and 1919, there was an epidemic of an influenza-like illness starting in Austria and France and spreading throughout Europe and North America. The illness was characterized by fever, headache, lethargy, and paralysis, particularly of the extraocular muscles. Following this, stupor, coma, sleep disturbance, and seizures could occur. Ocular gyric crises were seen in a high proportion of patients. Mortality was 30% to 40%, and parkinsonism developed in the majority of survivors over the next 10 years.158,159 The specific agent causing encephalitis lethargica was never identified. Although there has been no outbreak of encephalitis lethargica since the 1920s, infection as a cause for PD has still attracted some attention. There are numerous anecdotal reports of infections, particularly encephalitis, being associated with parkinsonism. These include a wide variety of viruses, bacteria (including Borrelia burgdorferii [Lyme disease]), and even fungi, such as Cryptococcus or Aspergillus. However, there is no evidence to suggest that any of these are relevant to the vast majority of patients with idiopathic PD. For instance, patients who develop PD before the age of 40 have no greater history of central nervous system infection than do patients who develop the disease over the age of 60. Intrauterine exposure to, for instance, the influenza viruses pandemic from 1890 to 1930 has not been supported by any association with year of birth.160 Searches for viral particles of antigens within the brains of patients with PD have not proved rewarding.161
Two environmental factors are recognized to lower the risk for PD: cigarette smoking162 and coffee drinking.163 The mechanisms through which they can reduce risk are not known. Coffee drinking appears more protective for men, so it is possible that there is an interaction with endocrine factors. There is evidence of active inflammatory change in the substantia nigra at the time of death in PD, with microglial activation, and expression of proinflammatory cytokines.164–166 The role of this inflammatory change is unknown but has been believed to be relevant to pathogenesis and neuronal damage. Similar changes have been seen in AD brain and prompted retrospective analyses that subsequently demonstrated the potential for antiinflammatory agents to reduce the risk for AD, although this effect remains controversial.167,168 A similar study in PD has also shown that use of a nonsteroidal antiinflammatory drug two or more times per week can produce a 45% lower risk for PD.169
PATHOGENESIS
Iron
High iron concentrations are found in control substantia nigra, globus pallidus and striatum. In PD, there is a 35% increase in substantia nigra iron levels.170,171 Other degenerative diseases involving cell loss in the basal ganglia also showed increased iron in these areas, such as progressive supranuclear palsy and multiple system atrophy. These studies suggested that increased iron concentrations were a reflection of neuronal cell loss rather than any specific pathogenetic factor. High concentrations of iron were also found in macrophages, astrocytes, and reactive microglia in the PD substantia nigra.172 One study, however, using x-ray microanalysis, found increased levels of iron in neuromelanin. In this respect, neuromelanin could again act as a toxic sink.173 In contrast, another study found no difference in iron concentrations between melanized and nonmelanized cells in controls but a significant increase in the cytoplasm of dopaminergic neurons.174 However, there was no apparent correlation between the high concentrations of iron and morphological alterations in the neurons that might suggest degeneration.
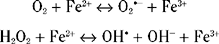
Thus, if iron is available in a free and reactive form, it has the potential for exacerbating oxidative stress and damage. Iron is normally bound to ferritin, which exists in two forms, H and L. Most brain ferritin is in the H form. Three studies have now been undertaken on ferritin concentrations in PD brain. One used a polyclonal antibody predominantly against L-ferritin and found a significant decrease in the concentration of this protein in PD substantia nigra and other areas.175 This decrease was not seen in other parkinsonian syndromes such as progressive supranuclear palsy or multiple system atrophy. Another study,176 again using an antibody against mainly L-ferritin, found an increase in the number of ferritin-positive microglia in substantia nigra. This latter work used immunohistochemistry and therefore is not directly quantifiable. In addition, this study incorporated parkinsonian syndromes as well as idiopathic PD into the disease group (M. Youdim, personal communication). A third and more comprehensive study involved monoclonal antibodies against both L and H ferritin together with a double capture technique incorporated into an enzyme-linked immunosorbent assay study together with Western blotting studies of PD substantia nigra protein.177 The results did not identify any significant difference in ferritin levels between control and PD substantia nigra. Thus, there are no hard data that ferritin levels are abnormal in PD. Indeed, ferritin has such a high iron binding capacity that the increase of iron noticed in PD brain may not require any increased buffering capacity from ferritin.
Oxidative Stress and Damage
There are several lines of evidence that suggest increased oxidative stress and oxidative damage to biomolecules in PD substantia nigra (Table 71-3).
TABLE 71-3 Oxidative Damage in Parkinson’s Disease
The free radical gas nitric oxide (NO•) is present in many tissues, including the central nervous system. NO• is generated by the conversion of L-arginine to L-citrulline by nitric oxide synthase (NOS). At least three NOS isoforms are recognized and are all expressed within the brain. NO• acts as an atypical molecular messenger but at higher concentrations may have a toxic role and be implicated in the neurodegeneration that occurs in PD. As a free radical, NO• could potentially contribute to dopaminergic neuronal death by mechanisms such as increased lipid peroxidation, release of iron(II), and damage to DNA. It can also inhibit a number of enzymes such as cytochrome c oxidase and superoxide dismutase and affects mitochondrial function by inhibiting complexes II, III, and IV. Animal studies have implicated NO• in nigrostriatal neuronal loss. In addition to its possible neuroprotective effect with regard to 1-methyl-4 phenylpyridinium (MPP+) toxicity, 7-nitroindazole (7-NI) also protects in the methamphetamine animal model of PD.186 NOS activity is at its highest in the nigrostriatal system in nonhuman primates and humans. Attempts to demonstrate altered levels of NO• in the brains of PD patients have been inconclusive with both decreased and increased levels of cerebrospinal fluid nitrate, a marker of NOS activity, being seen.187–189
Mitochondrial Dysfunction
The first link between mitochondria and PD was made in 1989 when a defect in the activity of respiratory chain protein complex I was identified in substantia nigra from patients with PD.190,191 This study has been expanded over the years, and results to date show that there is a specific defect of approximately 35% complex I deficiency in PD nigra.177 This defect in complex I activity in PD brain does not affect any other part of the respiratory chain. In addition, no defect in mitochondrial activity has been identifiable in any other part of PD brain, including the caudate putamen, globus pallidus, tegmentum, cortex, cerebellum, and substantia innominata.192
Following the report of complex I deficiency in PD substantia nigra, respiratory chain abnormalities were described in skeletal muscle mitochondria from PD patients. This particular area has proved very contentious, with several groups either describing similar defects or no abnormality whatsoever (see Schapira193 for review). Two magnetic resonance spectroscopy studies on skeletal muscle mitochondrial function in PD have shown conflicting results.194,195 Finally, mitochondrial complex I deficiency was also identified in platelet mitochondria of PD patients.196–198 In contrast to skeletal muscle, there is a consensus among several laboratories that complex I deficiency does exist in PD platelet mitochondria. The majority of studies, however, suggest that this deficiency, as least based on a group-to-group analysis, is modest (about 20% to 25%) (see Schapira193 for review). The complex I deficiency in PD lacks the sensitivity to allow its use as a biomarker of PD.
CLINICAL FEATURES
Presymptomatic
Olfactory dysfunction is common in PD and eventually affects up to 90% of patients.199 It has been suggested that hyposmia may be a preclinical marker for PD,200,201 and olfactory deficits have been reported in asymptomatic relatives of patients with PD, some of whom subsequently developed PD.202,203 A prospective study involving 361 asymptomatic relatives of PD patients identified 40 with hyposmia.9 Within 2 years of follow-up, 10% of this subgroup had developed PD and another 12% had detectable presynaptic abnormalities on their dopamine transporter SPECT scan. No relative with normal smell had an abnormal SPECT scan or developed PD. It is tempting to relate these findings to Braak’s findings of Lewy body distribution in the olfactory nucleus in stage 1. Olfactory dysfunction has also been reported in diffuse Lewy body disease and multiple system atrophy. Vascular parkinsonism, cortocobasal degeneration, progressive supranuclear palsy, and parkin-associated PD usually have intact olfactory function.204–206
Rapid eye movement (REM) behavior disorder (RBD) is a common sleep disorder in PD. It is characterized by loss of the normal skeletal muscle atonia during REM sleep, thus enabling patients to physically enact their dreams, which are often vivid or unpleasant.207–210 Vocalizations (talking, shouting, vocal threats) and abnormal movements (arm/leg jerks, falling out of bed, violent assaults) are commonly reported by bed partners. In PD, up to one third of patients meet the diagnostic criteria of RBD.207,208 RBD appears to frequently precede the development of motor signs of PD and longitudinal data that suggest that RBD heralds the onset of motor symptoms in up to 40% of PD patients.211,212 In patients with isolated RBD, imaging studies have indicated a small but significant reduction in striatal dopaminergic uptake that may suggest preclinical PD.213,214 The anatomical basis of RBD is believed to involve the pontomedullary area resulting from degeneration of lower brainstem nuclei like the pedunculopontine and subceruleal nucleus; this area is consistent with Braak stages 1 and 2.
Constipation often precedes the diagnosis of PD.215–218 A prospective study of 7000 men for 24 years assessed inter alia for bowel habits found that those with constipation (less than one bowel movement per day) had a threefold risk of subsequently developing PD.219 The mean interval between the administration of the bowel questionnaire and the development of PD was 10 years. Colonic dopaminergic neurons degenerate with Lewy body formation in PD, although constipation does not respond well to dopaminergic treatment.220–222
Motor Features
Rigidity represents an increase in tone that is present throughout the range of movement and is independent of the speed at which the limb is moved. The tremor of PD may superimpose on rigidity to produce cog-wheeling, and this phenomenon may be absent if there is no tremor. Examination of the wrist with gentle flexion-extension movements is the best means to elicit cog-wheeling, and this can be repeated at the elbow. Rigidity affects the patient’s posture, producing a flexion at most joints including the spine, and this produces the simian posture typical of PD. An extreme form of this is known as camptocormia.223 Postural abnormalities also affect the distal limbs with extension of the fingers and flexion of the metacarpophalangeal joints or dorsiflexion of the great toe (striatal hand or toe).
The development of an asymmetrical intermittent resting tremor at 4 to 6 Hz is estimated to be a manifesting feature in 70% of PD patients. In addition, there is often a higher frequency (about 12 Hz) small-amplitude postural tremor.224 The resting hand tremor is referred to as “pill-rolling” in the style of the pharmacists of the nineteenth century, who would prepare their tablets by hand. A tremor may affect other parts including the foot or leg (in which it may first manifest), lips, jaw, and tongue.225 A head tremor, titubation, is more suggestive of essential tremor. The resting tremor is exacerbated by physical or emotional stress and can in the early stages be voluntarily inhibited for short periods. The tremor usually becomes bilateral after about 5 to 6 years, although the first affected side most often remains the more severe.226
Nonmotor Features
The widespread and progressive neurodegeneration in the PD brain leads to the emergence of a variety of features that are collectively grouped under the title of nonmotor symptoms. These are predominantly, but not exclusively, the consequence of loss of nondopaminergic pathways (Table 71-4). The nonmotor symptoms of PD range from cognitive problems such as apathy, depression, anxiety disorders, and hallucinations to fatigue, gait and balance disturbances, hypophonia, sleep disorders, sexual dysfunction, bowel problems, drenching sweats, sialorrhea, and pain. These symptoms are often the most troubling for patients and contribute significantly to morbidity and impaired quality of life.227 Diplopia is a frequent symptom even in early PD, although the neurological basis is not known.
TABLE 71-4 Symptoms Less Responsive to Dopaminergic Therapy
| Motor | Postural instability |
| Gait disorders | |
| Speech problems | |
| Mental changes | Dementia |
| Depression | |
| Anxiety | |
| Apathy | |
| Autonomic nervous system dysfunction | Orthostatic hypotension |
| Constipation | |
| Sexual dysfunction | |
| Urinary problems | |
| Sweating | |
| Sensory phenomenon | Pain |
| Dysesthesias | |
| Sleep disturbances | Sleep fragmentation |
| Sleep apnea | |
| REM behavioral disorder |
Abnormalities of sleep are common in PD and are the result of a combination of the natural consequences of aging, the underlying disease pathology,228 motor and nonmotor complications,229,230 and drugs.231 Disordered sleep often results in excessive daytime sleepiness, and this in turn may be compounded by the sedative effect of dopaminergic drugs.232 Excessive daytime sleepiness and involuntary dozing affects up to 50% of PD patients and may be preclinical markers.233 In some, excessive daytime sleepiness has been linked to the development of sudden onset of sleep and a pattern reminiscent of narcolepsy with abnormal sleep latency period (<5 minutes) in 30% of PD patients. Polysomnographic studies have showed transition from wakefulness to sleep stage 2 within seconds without the sudden onset of REM sleep.234,235 Following reports of road traffic accidents caused by “sudden irresistible attacks of sleep” in eight PD patients, a large body of research focused on the possible effects of dopaminergic drugs and disease progression and the occurrence of sudden onset of sleep.232,236–238 The issue remains unclear, although is now regarded as part of the nonmotor complex of the disease progression in PD.239
Sexual and bladder dysfunction is common and occurs in both sexes. The dopaminergic treatment of PD may lead to increased sex drive, but the effects of the disease often result in impaired sexual performance.240 Bladder abnormalities particularly cause problems at night with nocturia, which when associated with bradykinesia during nocturnal “off” causes considerable discomfort.
Pain is a frequent symptom in PD, and some patients present especially with shoulder pain. Pain, anxiety, akathisia, respiratory distress, depressive mood swings, and slowed and impaired thought are symptoms that may be experienced during “off” periods and that will respond, at least in part, to dopaminergic therapy.241–243
DIFFERENTIAL DIAGNOSIS
The diagnosis of PD is best predicted by the presence of an asymmetrical bradykinetic rigid syndrome with a resting tremor and a good response to levodopa.244 The diagnostic specificity of these criteria is estimated at 98.6%, and sensitivity, at 91.1%.245 However, many patients still present a diagnostic challenge, especially those who have no tremor or those with marked asymmetrical tremor but limited bradykinesia or rigidity246 (Tables 71-5 and 71-6). Imaging studies on early PD patients recruited into neuroprotection trials indicate that approximately 10% have normal PET or SPECT scans.247,248 The Parkinson-plus disorders, dementia with Lewy bodies, Wilson’s disease, and tremor are covered in Chapters 72, 70, 108, and 33, respectively, and some of the additional diseases that may mimic PD have been discussed earlier. The clinical features of the main disorders that require differentiation from PD are covered only briefly in the context of diagnosis (see Table 71-4).
TABLE 71-5 Differential Diagnosis of Parkinson’s Disease
TABLE 71-6 Clinical Features of Parkinsonian Disorders
| Multiple System Atrophy | Corticobasal Degeneration | Progressive Supranuclear Palsy |
|---|---|---|
Drug-Induced Parkinsonism
Any dopamine receptor blocker has the potential to induce parkinsonism. In practice, the most common causes are the major neuroleptics, but drugs such as metoclopramide can also induce symptoms that may be confused with PD. A drug history is essential when considering PD. Clinical clues for drug-induced parkinsonism include symmetry and the absence of a resting tremor, although their presence does not exclude a drug cause. The presence of akathisia and orofacial dyskinesias supports a drug cause.249 Withdrawal of the drug may result in remission, although this can take months or years, and in some patients, the parkinsonism and related movement disorders are permanent.
Essential Tremor
Typical essential tremor comprises a bilateral usually symmetrical, visible, and persistent upper limb postural or kinetic tremor.250 Bradykinesia, rigidity, and postural abnormalities are not present. The tremor of essential tremor is present at rest in only 10% of cases251 but, when present or when asymmetrical, can cause difficulty with a distinction from PD, although such patients usually evolve to PD.252 The presence of a head or voice tremor, a strong and usually autosomal dominant family history, and improvement with alcohol all favor a diagnosis of essential tremor. In contrast, clear asymmetry, the presence of bradykinesia or rigidity, and leg tremor support a diagnosis of PD.
Multiple System Atrophy
Multiple system atrophy is a multicentric neurodegenerative disease that includes degeneration of the SNc and hence clinical features that can mimic PD. However, multiple system atrophy patients exhibit additional symptoms that may be predominantly cerebellar or parkinsonian with autonomic failure.253 Onset is similar in age to PD. The diagnosis of multiple system atrophy is supported by the presence of early cerebellar signs, autonomic failure such as bladder dysfunction or postural hypotension, early falls, pyramidal signs, myoclonus, bulbar features, pronounced anterocollis, and a rapid course. The response to levodopa is usually limited in degree and time,254 although some patients can develop dyskinesias, particularly of the orofacial and cervical musculature; they are rare.255
Progressive Supranuclear Palsy
Progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) typically manifests with gaze palsy, particularly of the vertical downward plane, a staring appearance, symmetrical parkinsonism, and pseudobulbar palsy.256 Axial greater than limb rigidity and falls within the first year are suggestive of progressive supranuclear palsy.257 Response to dopaminergic therapy is poor.
Corticobasal Degeneration
Corticobasal degeneration usually manifests as an asymmetrical disorder that may include abnormal limb posturing (alien limb), dystonic upper limb posturing with irregular jerks, cortical sensory deficits, primary progressive aphasia, apraxia, and parkinsonism. Tremor is uncommon; cognitive disturbance, particularly involving frontal lobe function, is often seen and frequently is the manifesting sign.258 The disease is rapidly progressive, and there is no or only a very poor response to levodopa.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


