Paroxysmal Disorders
Raman Sankar
Susan Koh
Joyce Wu
John H. Menkes
This chapter discusses conditions manifested by sudden, recurrent, and potentially reversible epileptic alterations of brain function.
EPILEPSY
Epilepsy was known to the ancient Babylonians and was described by Hippocrates, who considered it a disease of the brain. Its history, related by Tempkin, spans that of medicine itself (1). Hughlings Jackson concisely defined epilepsy as “an occasional excessive and disordered discharge of nerve tissue” (2). More recently, epilepsy has been defined as recurrent convulsive or nonconvulsive seizures caused by partial or generalized epileptogenic discharges in the cerebrum.
The epilepsies represent a group of diseases for which recurrent seizures represent their principal manifestation.
Estimates of the incidence of epilepsy depend on whether a single convulsive or nonconvulsive episode and febrile seizures are included in the definition. According to Millichap, febrile seizures account for 2% of all childhood illnesses (3). More recent estimates of the prevalence of single and recurrent nonfebrile seizures in children younger than 10 years of age range from 5.2 to 8.1 per 1,000 (4,5). By age 40 years, the cumulative incidence is 1.7% to 1.9% (4,5).
Classification
The epilepsies have been designated as primary (idiopathic), secondary (symptomatic), or reactive (Table 14.1). The term primary implies that, with the present knowledge, no structural or biochemical cause for the recurrent seizures can be found. In general, the primary epilepsies are genetically transmitted, and they tend to have a better prognosis for seizure control. The term secondary (symptomatic) epilepsy indicates that the cause of the seizure can be discovered. Such seizures are the principal manifestation of many diseases. They occur in the course of many congenital or acquired conditions of the nervous system, or they can complicate systemic disease. The designation of an epileptic condition as cryptogenic implies that the underlying etiology is symptomatic, but not readily demonstrable by available diagnostic techniques (6). In the reactive epilepsies, seizures are the consequence of an abnormal reaction of an otherwise normal brain to physiologic stress or transient insult. A notable example is febrile seizures. Not all epilepsies can be categorized conveniently. Some are atypical, others are rare, and for a significant proportion data necessary for classification are inadequate or incomplete.
The characteristics for all epilepsies are recurrent convulsive or nonconvulsive seizures. The 1989 classification scheme of the International League Against Epilepsy (ILAE) elected a hierarchy of dichotomies in which the initial categorization is based on whether the epilepsy is localization-related or generalized (6). This distinction was, in fact, made by Hughlings Jackson more than 100 years ago (7) (Table 14.2). Localization-related epilepsies (partial or focal) seizures are classified into simple, complex, and secondarily generalized. Simple partial seizures involve preserved consciousness, whereas complex partial seizures are those with impaired consciousness. The prevalence of the various seizure types is presented in Table 14.3.
The descriptive classification of epileptic syndromes is extremely useful clinically. The so-called epileptic syndromes are distinctive in that they demonstrate characteristic age of onset, seizure types, electroencephalographic (EEG) features, and prognosis. This is particularly valuable in pediatric epileptology because the immature brain often produces stereotypic epileptic behaviors that are a function of its stage of development, rather than etiology. Childhood syndromes can be considered as benign or catastrophic based on their responsiveness to treatment, the possibility of remission of seizures, and the long-term prognosis for normal cognitive development.
Etiology
Recurrent seizures are thought to result from a genetic predisposition, underlying neuropathologic changes, and chemicophysiologic alterations in the nerve cell and its
connections. Each of these factors is considered in turn. Attributed causes for epilepsy in children and adolescents are presented in Table 14.4 (8).
connections. Each of these factors is considered in turn. Attributed causes for epilepsy in children and adolescents are presented in Table 14.4 (8).
TABLE 14.1 Scheme for Organizing Epileptic Conditions | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
TABLE 14.2 Classification of Epileptic Seizures | ||
|---|---|---|
|
Genetic Factors
Numerous studies suggest that the genetic susceptibility to seizures is normally distributed in the general population, and that there is a threshold above which the condition becomes clinically evident.
An interaction between one or more genes and various nongenetic events operates in several conditions accompanied by seizures. These include head trauma, brain tumors, and congenital hemiplegia (9,10). Genetic factors appear to be most significant in patients with the various primary epilepsies (11). The various nonprogressive hereditary epilepsies are summarized in Table 14.5 (12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30). In one study, Lennox and Lennox found a 70% concordance for monozygotic twins and 5.6% concordance for dizygotic twins for epilepsies without organic brain lesions (31). Metrakos and Metrakos found a 12% incidence of seizures among parents and siblings of children with absence seizures; 45% of siblings had an abnormal EEG. They proposed that this EEG abnormality is an expression of an autosomal dominant gene with nearly complete penetrance during childhood and low penetrance in infancy and adult life (32). Gerken and Doose, interpreting data derived from their clinic, concluded that it was unlikely that a single autosomal dominant gene was responsible for the 3-Hz spike and wave trait and suggested a polygenic inheritance with neurophysiologic and genetic heterogeneity (33). Indeed, at least three genes for absence epilepsy have been mapped at this point in time (see Table 14.5) (34,35).
TABLE 14.3 Prevalence Rates Per 1,000 of Specific Seizure Types in Children Aged Newborn to 9 Years | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
In families with centrotemporal spikes or sharp-wave discharges and rolandic seizures, EEG abnormalities are transmitted in a dominant manner with age-dependent penetrance (11). Only 12% of relatives with EEG abnormalities, however, develop clinically apparent seizures. This type of seizure has been mapped to chromosome 10q22-q24, with the defective gene being LGI1 (36). A significant genetic predisposition also occurs in juvenile myoclonic epilepsy, in photosensitive seizures, and in the various other primary generalized epilepsies. In seizures
with secondary generalization, the genetic factors, although demonstrable through controlled twin studies, are not as striking as in the primary generalized epilepsies. However, even in absence epilepsy, in which the genetic factor is most prominent, the overall risk of developing seizures is only 8% for siblings of affected subjects and 2% in as yet unaffected siblings older than 6 years of age (37). For offspring of subjects with absence seizures, the risk for EEG abnormalities is 64% and for seizures is 6.7% (38). When promazine is used to activate the EEG, 73.5% of 7- to 14-year-old siblings of subjects with idiopathic absence seizures develop an abnormal EEG (39).
with secondary generalization, the genetic factors, although demonstrable through controlled twin studies, are not as striking as in the primary generalized epilepsies. However, even in absence epilepsy, in which the genetic factor is most prominent, the overall risk of developing seizures is only 8% for siblings of affected subjects and 2% in as yet unaffected siblings older than 6 years of age (37). For offspring of subjects with absence seizures, the risk for EEG abnormalities is 64% and for seizures is 6.7% (38). When promazine is used to activate the EEG, 73.5% of 7- to 14-year-old siblings of subjects with idiopathic absence seizures develop an abnormal EEG (39).
TABLE 14.4 Attributed Causes for Epilepsy in Children and Adolescents by Sex: Prevalent Cases, 1983 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 14.5 Genetically Transmitted Nonprogressive Epilepsies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Several other genes responsible for epilepsy have been mapped and cloned. The first epilepsy gene to be cloned was one of three genes responsible for autosomal dominant nocturnal frontal lobe epilepsy (24,40). It has been mapped to chromosome 20q13.2-13.3 and encodes the nicotinic acetylcholine receptor alpha-4 subunit (CHRNA 4). The same authors later reported a different mutation in the same gene for a different pedigree with this syndrome (41). Two other genes for this condition have been mapped to chromosome 15q24 and chromosome 1 (24). The discovery of mutations in this gene was perplexing to many because this receptor has not been considered to be involved in the modulation of neuronal excitability relevant to seizure disorders.
The finding that benign familial neonatal convulsions are attributable to mutations of voltage-gated potassium channels, KCNQ2 (12,13,42,43) and KCNQ3 (44), is more in tune with our understanding of the mechanisms of excitability. Altered K+-channel function could impair neuronal repolarization and thus contribute toward increased excitability. The extremely transient nature of this disorder suggests that compensatory changes probably take place in other genes controlling excitatory or inhibitory ion channels.
The relationship between the genotype and the phenotypic expression of the gene disorder is complex and is complicated by phenotypic convergence—that is, two different genetic mutations can induce the same clinical picture. Thus, afebrile seizures during the first year of life can result not only from mutations in the sodium channel gene, SCN2A1, but also from mutations in the GABA receptor gene, GABRG2. Conversely, there is phenotypic divergence, and different mutations of the same calcium channel gene CACNA1A are associated with familial hemiplegic migraine, episodic ataxia, and epilepsy (45). Mutations in LGI1 can be associated with a variety of phenotypes: partial epilepsy with auditory features, mesial temporal lobe epilepsy, temporal lobe epilepsy with febrile seizures, and temporal lobe epilepsy with developmental delay (36). Mutations in SCN1A are associated with a clinical continuum, including severe myoclonic epilepsy of infancy, generalized epilepsy with febrile seizures plus, and intractable childhood epilepsy with tonic-clonic seizures as well milder forms such as classical “febrile seizures”(46). Likewise, mutations in the chloride channel gene CLCN2 can be found in childhood absence epilepsy, juvenile myoclonic epilepsy, or epilepsy with grand mal upon awakening (46a). The reasons that underlie this clinical diversity are unclear. It is likely that additional genes contribute and modify the phenotypic expression. Indeed, Durner and colleagues found statistical support for a major susceptibility gene for idiopathic generalized epilepsy and different modifying genes (47).
Neuropathologic Factors
Seizures can occur in patients with almost any pathologic process that affects the brain. Two types of abnormalities are seen: those that are responsible for recurrent seizures, and those that are the consequence of recurrent seizures.
Gowers stated more than 100 years ago that seizures beget seizures (48). The question whether lesions produce seizures or seizures produce lesions has been extensively investigated.
Lesions Responsible for Recurrent Seizures
A variety of morphologic changes can cause recurrent seizures. They range from the most obvious, such as some of the major developmental anomalies (see Chapter 5) or postasphyxial changes (see Chapter 6), to minor dysgenetic lesions such as the gray matter heterotopias (see Chapter 5). Although morphologic alterations would not be expected to be found in the primary epilepsies, sometimes they are (49). Mutations in the gene filamin A, that codes for a protein with a role in actin cross-linking and membrane stabilization have been reported to be responsible for the aberrance in migration that results in periventricular heterotopia (50,51). A number of authors have called attention to minor developmental anomalies in the molecular layer of the cerebral cortex and in the cerebellar cortex, some of which are clearly the result of disturbed cell migration (52). Malformations, notably gray matter heterotopias, cryptic tubers, or angiomas arising within the temporal lobe, can cause recurrent seizures. Such lesions also can be found in other areas of the brain (53,54). The genetic basis of some of the dramatic cerebral malformations associated with severe epilepsies of early childhood, such as the double cortex syndrome or band heterotopia, are also beginning to be understood (55,56) (see Chapter 5).
Altered neuronal migration that results in granule cell disorganization in the dentate gyrus has been seen in tissue resected from patients with temporal lobe epilepsy (57). Although initially this was thought to be a congenital lesion, provocative data from Parent demonstrates that, even in mature animals, status epilepticus can result in neurogenesis in the dentate gyrus, and that the nascent granule cells may migrate aberrantly. The data suggest that aberrant
synapse formation by these cells could contribute to abnormal excitability (58,59,60).
synapse formation by these cells could contribute to abnormal excitability (58,59,60).
The role of infectious processes in the pathogenesis of epilepsy has received relatively little attention since Aguilar and Rasmussen established that some epileptic patients with focal seizures, slowly progressive intellectual deterioration, and cerebral atrophy demonstrate a pathologic picture consistent with a viral encephalitis (61). Attempts at viral isolation have been unsuccessful in these cases. Nevertheless, it is likely that not only focal epilepsy, but also other forms of seizure disorders, particularly disorders beginning in early childhood in previously healthy children (such as epileptogenic encephalopathy or progressive facial hemiatrophy), are caused by a smoldering viral disease within the brain (see Chapter 7).
Lesions Secondary to Recurrent Seizures
Among the lesions considered to be secondary to recurrent seizures are those that result from the physical trauma that often attends seizures, and those that result from hypoxia, vascular alterations, or the action of the excitatory neurotransmitters.
Meldrum and colleagues (62) explored the possibility that the seizure itself, rather than systemic changes, was responsible for brain damage. They showed that brain damage occurred in the absence of systemic abnormalities in paralyzed, ventilated, adolescent baboons that were subjected to prolonged, bicuculline-induced seizures. Although the neurochemical changes attending cell death owing to prolonged seizures are similar to those seen in ischemia and hypoglycemia, significant differences in the time course and anatomic distribution of brain damage occur (see Table 17.1).
Under clinical conditions, damage results from a combination of the increased metabolic demands that accompany excessive neuronal activity and the reduced circulation and substrate supply induced by the combination of hyperthermia, hypoglycemia, hypotension, and hypoxia. Cell death under these conditions occurs through a process that resembles cell death in asphyxia, namely through the release of excitotoxins that increase intracellular calcium in the course of prolonged seizures. A more extensive discussion of this process can be found in Chapter 6.
TABLE 14.6 Possible Etiologic Factors for Complex Partial Seizures | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
Several areas of the brain, especially the hippocampus, appear to be particularly vulnerable to recurrent and prolonged seizures. Anatomic manifestations of cell damage to the hippocampus include loss of interneurons in the hilus, pyramidal cell loss within the Sommer’s sector (prosubiculum and subfield CA1 of Ammon’s horn), and subfield CA3, with consequential glial scarring and atrophy (63,64). Using Golgi techniques to study the hippocampus and dentate nucleus, Scheibel and associates have observed loss of dendritic spines and deformation of the dendritic shaft (65). This selective hippocampal vulnerability has been postulated to result from a high density of excitatory receptors on nerve cells in Sommer’s sector (66). Other factors also could be operative. Using in situ hybridization techniques, Sommer and coworkers showed unique developmental patterns in the mRNA expression of the Glu R-1, -2, and -3 glutamate receptor subunits in CA1, CA3, and the dentate gyrus. Differences in receptor structure could result in differences in receptor function and differences with maturation in resistance of the hippocampus to epileptic damage (67). The protective role of calbindin, a calcium-binding protein, from glutamate-induced neurotoxicity also could account for the selective nerve cell loss (68).
Within the gray matter of the cerebral hemispheres, neuronal cell loss is most likely to occur in laminae 3 and 4, where the thalamocortical afferents terminate. Damage also occurs in the pars reticularis of the substantia nigra, globus pallidus, and thalamus. In animal models, the substantia nigra has been demonstrated to play an
important role in the propagation of seizures, and damage to this structure could conceivably contribute to increased propensity for seizures (69). The caudate nucleus appears to be spared (70).
important role in the propagation of seizures, and damage to this structure could conceivably contribute to increased propensity for seizures (69). The caudate nucleus appears to be spared (70).
There is controversy as to the effect of seizures on the immature brain and whether brief but repetitive seizures can induce brain damage. One of us (R.S.) has reviewed the arguments for both sides in this controversial issue (71,72). Some types of experimental seizures fail to produce histologic lesions (71). This observation gave rise to the argument that the immature brain is not vulnerable to seizure-induced damage. This argument runs contrary to the observations on surgically resected tissue from epileptic children that show structural alterations that can be attributed to seizures (73,74). More recent work has shown that the effect of seizures on the developing brain is age- and model-specific, and also that both prolonged and brief and repetitive seizures are associated with the induction of neuronal apoptosis in specific cell populations in human and experimental animals (75,76,77,78).
Brief but recurrent seizures induced by pentylenetetrazol have been shown to contribute to morphologic and functional alterations in neonatal rat pups (79,80,81). The question whether brief but recurrent seizures also have a similar potential to induce brain damage cannot be answered with assurance in humans. In terms of clinical practice, patients with recurrent seizures are invariably treated with antiepileptic drugs, which on their own can affect development (82); the natural history of the condition without treatment is, therefore, impossible to study. Autopsy material does not permit an easy distinction of the pathology that caused the frequent seizures from the effect of the seizures themselves.
The studies of Shewmon and Erwin are more directly applicable to the clinical problem. These workers have demonstrated that interictal spikes, when followed by prominent inhibitory after potentials, can transiently disrupt cortical function. Thus, frequent interictal spikes could interfere with modality-specific learning (83). Presumably, recurrent electrical discharges could influence activity-dependent plasticity of the developing brain.
Mesial Temporal Sclerosis (Ammon’s Horn Sclerosis, Hippocampal Sclerosis)
The damage seen in the hippocampus obtained by surgical resection in chronic temporal lobe epilepsy differs from that seen in postmortem specimens after status epilepticus (84,85). In contrast to the selective damage seen after status epilepticus, the hippocampus of subjects with chronic temporal lobe epilepsy shows more widespread damage throughout the CA1, CA2, and CA3 subfields as well as the dentate granule cells (84). Hippocampal cell loss ranges from mild and random to almost complete. Although initially the changes have a bilateral distribution, with time, one hemisphere becomes more affected (86). This pathologic abnormality has been designated as mesial temporal sclerosis (MTS) or hippocampal sclerosis. It was seen in 47% of resected temporal lobes in the series of Falconer and associates (Table 14.6) (87), and in 64% of a more recent series compiled by Engel and associates (88). Cell loss and gliosis in the amygdala also has been observed and can occur in the absence of significant hippocampal changes (89). In more severe cases, nerve cell loss and gliosis involves not only the entire hippocampus, but also the uncus, amygdala, and adjacent cortex (Fig. 14.1). Atrophic changes in the cerebellum or the thalamus are not uncommon.
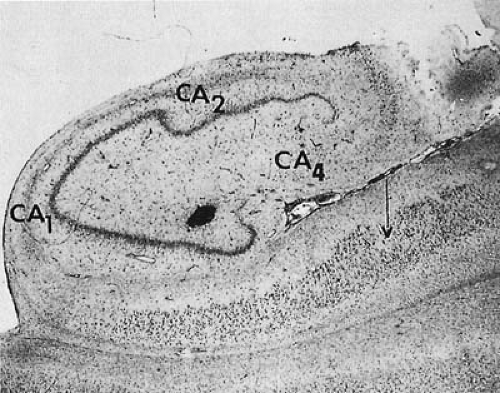 FIGURE 14.1. Mesial temporal sclerosis in 23-year-old man. Onset of complex partial seizures began at age 6 years, with a frequency of up to seven per day. A few major motor seizures occurred each year. Cross-section of the cornu Ammonis shows the extent of injury of the pyramidal cell layer. Neurons of areas CA4, CA2, and CA1 are markedly reduced in number. There are also focal areas of cell loss in the subiculum (arrow) (Gridley stain, ×10). (Courtesy of the late Dr. W. Jann Brown, Department of Pathology, University of California, Los Angeles, UCLA School of Medicine.) |
The cause or causes of MTS are still unresolved. MTS has been seen as early as 1 year of age, and combined morphologic and electrophysiologic studies suggest that the seizure focus is generated in part when abnormal, recurrent, monosynaptic excitatory synapses are formed after damage to normal intrahippocampal synapses (90). A detailed analysis of the epileptogenic potential of the lesions produced by intrahippocampal or systemic kainic acid or by ischemia led Franck to conclude that hippocampal sclerosis and seizures are both symptoms of an underlying pathology, and that although MTS may be produced by seizures, the development of epilepsy as a syndrome does not depend on cell loss or plasticity in the hippocampus (91). Neuroimaging studies performed within 48 hours of a prolonged febrile convulsion indicate the presence
of hippocampal edema that resolves within a few days, and that in some instances is replaced by hippocampal atrophy (92,92a). These studies do not resolve the questions of whether a pre-existing hippocampal abnormality predisposed to the prolonged febrile convulsion and whether the anatomic features of MTS may become incorporated into, and sustain, an epileptic focus. In this regard, the identification of an interleukin gene polymorphism identified with both prolonged febrile convulsions and temporal lobe epilepsy with hippocampal sclerosis is quite interesting (92b).
of hippocampal edema that resolves within a few days, and that in some instances is replaced by hippocampal atrophy (92,92a). These studies do not resolve the questions of whether a pre-existing hippocampal abnormality predisposed to the prolonged febrile convulsion and whether the anatomic features of MTS may become incorporated into, and sustain, an epileptic focus. In this regard, the identification of an interleukin gene polymorphism identified with both prolonged febrile convulsions and temporal lobe epilepsy with hippocampal sclerosis is quite interesting (92b).
Vascular, metabolic, genetic, and immunologic factors, acting singly or in concert, can be responsible for MTS (93,94). Epidemiologic studies have been used to ascertain risk factors for this condition. Febrile convulsions were seen in 20% of subjects, as compared with 2% of controls. The majority experienced at least one complicated febrile seizure. An increased incidence of head trauma and neonatal convulsions also could be documented. Additionally, there was a significant association with maternal seizures (95). In a significant population of patients, the brain, after a lifetime of recurrent epileptic attacks, shows neither gross nor microscopic abnormalities. This observation reflects the current limitation of morphologic studies in furthering our understanding of the epilepsies.
Basic Mechanisms of Epileptogenesis
We must notice what the normal function of nerve tissue is. Its function is to store up and expend force.
—H. Jackson, 1873 (96)
It should be stated at the outset that modulation of transmitter effects, of voltage-gated channels, and of cell electrical properties involves processes that occur continually during normal brain function. This plasticity is the basis of the cortex to learn from experience. It seems that the same plastic mechanisms are involved in epileptogenicity. One extreme result of such plasticity is the hyperexcitability and hypersynchrony that characterize epileptiform activities. The risk of epileptiform activity is the price that has to be paid for a nervous system that is so adaptive (97).
Each clinical form of epilepsy is generated by a different set of mechanisms. In general, there is greater understanding presently of generators of focal epileptiform activity than generalized epileptiform activities. Cellular aspects of epileptogenesis are reviewed by DeLorenzo (98) and Velísek and Moshé (99).
Neurophysiology and Neurochemistry
From a neurophysiologic point of view, an epileptic seizure has been defined as an alteration of central nervous system (CNS) function resulting from spontaneous electrical discharge in a diseased neuronal population of cortical gray matter or the brainstem.
Epileptogenesis requires a set of epileptogenic neurons, the presence of disinhibition, and circuitry to permit synchronization.
Partial Epilepsies
An epileptic neuron has, among other characteristics, an increased electric excitability and the ability to sustain an autonomous paroxysmal discharge that can be influenced from the outside by synaptic activity. Intracellular recordings within an epileptic focus reveal that during the time when an interictal discharge is recorded on the scalp EEG, a compact population of neurons displays a stereotyped abnormality called paroxysmal depolarization shift (PDS) in intracellular recordings. A PDS is characterized by a sudden, large, and sustained (approximately 30 mV for 70 to 150 msec) depolarization that is synchronized in many neurons. Multiple, high-frequency action potentials are superimposed on the PDS. The PDS and the interictal spike on scalp EEG, which represents that synchronized PDS of a local population of neurons can occur spontaneously or can be triggered by afferent stimuli. The PDS is followed by a hyperpolarization of 10 to 20 mV below the resting potential that lasts 700 msec or longer. During this period, the focus is refractory to afferent stimulation.
The large and lasting depolarization that characterizes a PDS is attributed to the triggering of voltage-gated calcium channels by the incoming action potential. This depolarizing calcium conductance is mediated by a subtype of excitatory amino acid receptor, which is characterized by its high affinity to N-methyl-D-aspartate (NMDA). The calcium channel is regulated by magnesium through a voltage-dependent block that can be removed by the initial sodium influx triggered by the action potential. The rise in intracellular calcium in turn triggers the opening of a specific type of potassium channel that initiates the hyperpolarization phase.
Ictogenesis is the spread of localized epileptic discharges to induce a clinical seizure during which thousands of neurons fire synchronously for prolonged periods. Several experimental systems have been used to study how localized discharges are able to spread. In a nonepileptic brain, an area of neuronal hyperpolarization, the inhibitory surround, surrounds the region of synchronous paroxysmal discharges. This inhibitory surround limits the duration of the interictal discharge, determines its frequency, and prevents its progression into a full-blown seizure. Neurons can become hyperpolarized by several processes that can differ from one set of neurons to another.
The mechanisms responsible for this transition from interictal to ictal period probably involve nonsynaptic processes such as electrical field effects (ephaptic interactions) and electrotonic coupling via gap junctions (100). Changes
in the extracellular environment, such as K+ and Ca2+ concentrations, also can affect the excitability of neuronal populations (101,102), suggesting that astrocytes also may play important roles in this process.
in the extracellular environment, such as K+ and Ca2+ concentrations, also can affect the excitability of neuronal populations (101,102), suggesting that astrocytes also may play important roles in this process.
The observation that the normal limbic system can become epileptogenic by repeated stimulation, a process termed kindling, has provided a model for the study of the development of complex partial epilepsy (103,104). Kindling refers to a process by which brief trains of subconvulsive electrical stimuli are repeatedly delivered at appropriate intervals to a susceptible area of the brain. Initially, these stimuli produce after-discharges, which become progressively more prolonged until they give rise to limbic and clonic motor seizures. When stimulations are continued for even longer periods, spontaneous seizures appear. Once established, the effects of kindling are permanent. Kindling also can be achieved by chemical stimulation of the cortex.
During kindling, the mossy fiber pathway (efferent from the granule cells of the dentate gyrus) undergoes reorganization of its synaptic connections (105). The resulting recurrent excitatory connections have been implicated in the progressive development of hypersynchronous discharge. Such synaptic reorganization associated with loss of pyramidal cells in the CA3 subfield has been demonstrated in human epileptic tissue (106,107,108). Sloviter has suggested that the recurrent mossy fiber terminals include synapses on inhibitory interneurons (109).
Using his perforant path stimulation model of status epilepticus, Sloviter has studied the development of mossy fiber sprouting, chronic epilepsy, and time-dependent alterations in dentate inhibition. He suggested that the recurrent mossy fiber terminals include synapses on inhibitory interneurons (109,110). In his conceptualization, the hilar basket cells [inhibitory, g-aminobutyric acid (GABA)-ergic] are deafferented by the loss of another group of cells, the mossy cells (excitatory, glutamatergic), which normally receive mossy fiber input, and drive the inhibitory basket cells (Fig. 14.2); hence the term dormant basket cell hypothesis for this concept. Mossy fiber sprouting compensates for the loss of drive to the basket cells. GABAergic cells, indeed, appear to be preserved in human epileptic tissue (111) and also in animal models (110), thus supporting this concept.
Studies have compared the expression of excitatory amino acid receptor subunits and glutamic acid dehydrogenase (GAD) (presynaptic marker for GABA terminals) to the extent of mossy fiber sprouting in tissue from patients who underwent surgery. Patients’ granule cell KA2 and GluR5 mRNA levels were increased in association with aberrant fascia dentata mossy fiber sprouting; however, increased glutamic acid dehydrogenase immunoreactivity also was present in such tissue (112,113).
The preceding discussions pertain to the structural (network) plasticity that may be associated with focal epileptogenesis. There also is evidence for functional plasticity of synapses. Excitatory synapses show robust enhancement when they undergo repetitive high-frequency activation (114). This includes facilitation during the course of sustained stimulation and long-term potentiation that lasts hours to days after such a burst of activity in excitatory synapses. In contrast, similar repetitive high-frequency driving diminishes the efficacy of inhibitory (GABAergic) synapses (115,116).
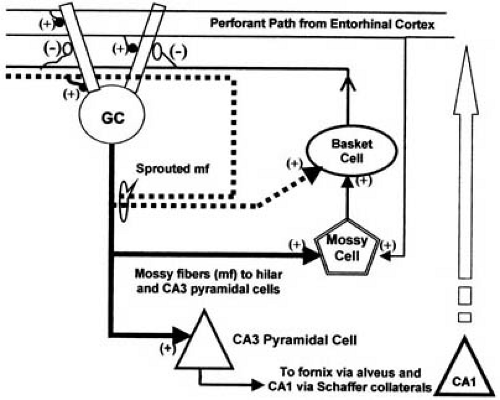 FIGURE 14.2. Hippocampal circuitry and seizure-induced circuit reorganization. Granule cells (GC) receive their major input via the perforant path. The perforant path also stimulates hilar interneurons (such as mossy cells and basket cells) to provide feed-forward inhibition of the granule cells. Granule cell axons, the mossy fibers, make synaptic contact with CA3 pyramidal cells. Mossy fiber collaterals innervate the hilar interneurons, such as the mossy cell shown in the diagram. Mossy cells are excitatory to GABAergic basket cells, which provide feedback inhibition to the granule cell. Sprouting of mossy fibers (in response to seizure-induced loss of CA3 pyramidal cells and hilar mossy cells) can result in enhanced excitation by forming autapses (an axon sprout synapsing with the dendrites of the same cell) and can augment synchronization by stimulating neighboring granule cells (not shown), thus contributing to epileptogenicity. It also has been suggested that the sprouted mossy fibers may restore inhibition lost after seizure-induced death of hilar mossy cells by direct stimulation of deafferented (dormant) basket cells. |
Evidence has emerged to suggest that the epileptogenic process also may involve cellular plasticity in addition to synaptic and network plasticity. Lasting changes in the subunit composition of hyperpolarization-induced, cyclic nucleotide-activated cation channels (HCN), which regulate cellular excitability, have been noted after hyperthermic convulsions in rodents (117). HCN channel plasticity also has been demonstrated in resected hippocampal tissue from humans (118). Alterations in channels that mediate low-threshold calcium currents (T-calcium channels) also has been demonstrated in both experimental and resected human hippocampi (119).
Three factors determine whether a focal seizure will become generalized. The first is the excitability of the epileptic neurons, the second is the ease with which an electric discharge can be propagated from the focus, and the third is the threshold of the brainstem centers for disseminating an electric discharge. The last is believed to reflect in part a genetic predisposition, and in part the frequency with which the brainstem centers are activated by the primary epileptic focus.
Current concepts propose that a secondarily generalized tonic-clonic seizure results from the axonal propagation of the cortical ictal discharge to the contralateral cortex, and to subcortical structures via intrahemispheric and interhemispheric association pathways. The substantia nigra, in particular, appears to be involved, at least in the control of experimental seizures (120).
Once neuronal excitation derived from the epileptic cortical focus spreads to involve the brainstem, particularly the midbrain and the pontine reticular formation, a generalized seizure develops almost instantly. These areas are responsible for the dissemination of epileptic potentials (121,122). With the subcortical neurons involved by the epileptic discharge, a positive excitatory feedback circuit is established between the cortex and the subcortical neurons, inducing discharges at a rate of 10 to 40 Hz. This circuit is responsible for the tonic phase of the focal motor seizure. As inhibitory neurons are recruited, a negative inhibitory feedback circuit develops, which periodically interrupts the excitatory activity and produces the clonic phase of the seizure. When the negative feedback wins ascendancy, the seizure subsides, leaving the neuronal membrane in a far greater hyperpolarized state than before the onset of the seizure.
Considerable evidence suggests that postictal (Todd’s) paralysis, a common sequel to a focal seizure, is caused by persistence of the active inhibitory state, rather than by metabolic exhaustion of epileptic neurons (123).
The various areas of the cortex differ in their potential for secondary generalization. A number of areas including the temporal, frontal, and prefrontal cortex have particularly strong corticofugal projections to the centrencephalic system, and focal lesions within them readily induce a generalized seizure discharge (124,125). By contrast, the potential for secondary generalization is low in the motor strip. Additionally, small cortical lesions are more likely to induce a focally restricted seizure, whereas multiple or diffuse cortical lesions are more likely to result in a generalized seizure.
Primary Generalized Epilepsies
The pathophysiology of the primary generalized epilepsies is less well understood, and much of the experimental data is based on animal models that might not be applicable to the human epilepsies. In the 1940s, Penfield introduced the concept of centrencephalic epilepsy. This idea was that a generalized spike and wave discharge, such as occurs in absence epilepsy, originates in the rostral brainstem structures and the diencephalon, with the thalamus being responsible for the sudden generalized cortical discharge (126).
Gloor and Fariello postulate that in the primary generalized epilepsies, the cortex is in a diffusely hyperexcitable state, perhaps as a result of the discharge of a group of excitatory neurons. As a result, an epileptic discharge can be triggered by excitation of the brainstem and the midline thalamic reticular system induced by thalamocortical input (127). Engel suggests that the firing of a small group of excitatory neurons stimulates a set of inhibitory neurons that have connections throughout the cortex. A second burst of properly timed excitatory impulses then produces a synchronized discharge over a wide area of the cortex (128). Thus, the spike-wave discharges arise from the rhythmic, reverberatory interactions between interconnected thalamic and cortical neurons.
The oscillations in the thalamocortical circuits rely in part on the intrinsic membrane properties of the involved thalamic neurons. These neurons undergo slow, calcium-dependent depolarizations, attributed to the so-called T-channel (129). These low-threshold calcium currents provide the pacemaker quality to these cells that forms the basis of the thalamocortical reverberations. These spike-wave paroxysms are abolished by substances such as trimethadione or ethosuximide, which have a specific effect of abolishing calcium currents associated with the T-channel (129).
Clinically, such a discharge produces grand mal or absence epilepsy. In the latter condition, quick excitation of a neuronal inhibitory system prevents a prolonged clinical seizure and the development of the tonic-clonic components. These seizures can occur without known cerebral injury or disease, and, as indicated previously, they have a high rate of genetic transmission.
Views on the propagation of cortical electrical discharges have been considerably modified in the last few years, and the importance of the cortex in the production of both primary and secondarily generalized seizures has become increasingly evident from implanted or surface electrodes and from ictal and interictal PET scanning. These techniques also have shown the marked discrepancies between the scalp EEG and the actual seizure focus in approximately one-third of patients with refractory seizures (130,131).
Excitatory and Inhibitory Neurotransmitters
Excitatory neurotransmitters can play a role in the development of seizure discharges. Glutamate and aspartate are the major excitatory neurotransmitters found in the
mammalian brain (132). Focal application of glutamate to hippocampal slices induces a calcium ion current and depolarizes the neuron. Another excitatory neurotransmitter system, the cholinergic system, has been successfully manipulated to produce experimental limbic seizures (133). Although the development of antiepileptic drugs based on their action at excitatory amino acid receptors is an active area of current research, presently available antimuscarinic agents are not likely to be useful as anticonvulsants because of their widespread central and peripheral sites of action.
mammalian brain (132). Focal application of glutamate to hippocampal slices induces a calcium ion current and depolarizes the neuron. Another excitatory neurotransmitter system, the cholinergic system, has been successfully manipulated to produce experimental limbic seizures (133). Although the development of antiepileptic drugs based on their action at excitatory amino acid receptors is an active area of current research, presently available antimuscarinic agents are not likely to be useful as anticonvulsants because of their widespread central and peripheral sites of action.
Nicotinic receptors have not been thought to be important in the mechanisms underlying seizures. The recent finding of an association between autosomal dominant nocturnal frontal lobe epilepsy and mutations in the gene for a nicotinic cholinergic receptor subunit (CHRNA4) was thus surprising to investigators (24).
GABA has been shown to have inhibitory postsynaptic activity and is one of the principal inhibitory neurotransmitters in the mammalian brain. The role of inhibitory GABA-releasing neurons in producing hyperpolarization has been well established, and temporary disinhibition might predispose a normal neuronal population to epileptiform activity. The GABA receptor has been found in all areas of the brain (134). This receptor is coupled to the chloride channel (chloride ionophore), so that GABA binding to its receptor results in a rapid opening of the chloride channel, with an ensuing increase in the postsynaptic membrane conductance to chloride. Increased chloride ion permeability stabilizes the cell near its resting membrane potential and reduces its response to excitatory inputs. Modulation of the GABA receptor-chloride ionophore complex mediates the actions of benzodiazepines and barbiturates as well as the convulsant effect of picrotoxin and its analogues (135). Interactions at the GABA receptor-chloride ionophore complex also underlie some of the mechanisms of action of felbamate and topiramate. These substances all have receptor sites on the GABA receptor complex. The receptor protein has been isolated and purified, and the genes coding for its five subunits have been cloned (136,137). The physiology and pharmacology of the receptor assembly depend on the subunit composition (137,138,139). No genetic diseases resulting from a defect in these genes have been demonstrated as yet.
Convulsions can be induced by substances that block the biosynthesis of GABA. Allylglycine, an inhibitor of glutamic acid decarboxylase, the enzyme promoting conversion of glutamic acid to GABA, is a potent epileptogenic compound (94). Inhibition of GABA binding to its receptor by bicuculline and inhibition of the postsynaptic GABA-chloride conductance responses by picrotoxin also induces convulsions. When GABAergic inhibition is progressively blocked with picrotoxin, stimulation of one neuron excites more and more neurons until neuronal behavior begins to resemble a seizure. Conversely, a number of compounds that elevate nerve-terminal GABA concentrations are potential anticonvulsants (140). These include such inhibitors of GABA transaminase as γ-vinyl-GABA (vigabatrin) and valproate. It is unclear, however, whether the anticonvulsant effects of valproate are indeed related to its elevation of brain GABA because this effect is seen only at supratherapeutic levels.
Magnetic resonance (MR) spectroscopy has provided evidence that gabapentin and topiramate also measurably increase brain concentrations of GABA even though they do not inhibit GABA transaminase (141,142). Extracellular (presumably also synaptic) concentration of GABA is increased by tiagabine, a nipecotic acid derivative (143).
Pyridoxine functions as a coenzyme for glutamic acid decarboxylase. Consequently, an increased glutamic acid to GABA ratio is expected in pyridoxine deficiency, a state marked by prolonged seizures. In pyridoxine dependency, an autosomal recessive disorder also characterized by seizures, the GABA content of brain is reduced, and brain glutamic acid concentration is elevated (144). Even though skin fibroblasts show reduced activity of pyridoxal-dependent glutamic acid decarboxylase, no abnormality in the two genes coding for glutamic acid decarboxylase, the biosynthetic enzyme for GABA, has been documented (145).
This relatively simple model for the epileptogenicity of insufficient inhibition and excessive excitation may, however, not hold true. Strong inhibition can predispose to hypersynchronization, and in petit mal seizures, hyperfunction of GABAergic inhibitory pathways is evident (146). Brainstem and diencephalic influences, which normally induce slow-wave sleep, also increase cortical neuronal synchronization and raise the potential for epileptogenicity. In some of the experimental epilepsies, notably the reflex epilepsy of Mongolian gerbils, the number of GABAergic neurons is increased (147).
A role for other inhibitory neurotransmitters, notably the opioid peptides, in the production of interictal inhibition, and a role for postictal depression have been suggested from various animal models (148). At low dosages, morphine and other opioids have anticonvulsant activity that is reversed by naloxone, whereas at higher dosages, they act as convulsants, producing a petit mal–like seizure disorder. The relevance of these observations to human petit mal is uncertain, although positron emission tomography (PET) has revealed increased opiate receptor binding in the temporal cortex in patients with complex partial seizures (149).
Adenosine is another potent inhibitor of cortical neurons, acting primarily by depressing spontaneous neuronal firing or synaptic transmission through its inhibition of the presynaptic release of excitatory neurotransmitters. In a rat model of limbic status epilepticus, an
adenosine A1 receptor agonist antagonized the development of status (150). Evidence from in vivo microdialysis experiences, performed intraoperatively on humans, suggests that adenosine may be a potential mediator of seizure arrest and postictal refractoriness (151). The clinical use of adenosine and of adenosine analogues has not been investigated extensively (152), and there is evidence that systemically administered adenosine analogues do not cross the blood–brain barrier (153). Moreover, the widespread effects of adenosine in the brain may not permit the development of a highly specific anticonvulsant with minimal CNS side effects.
adenosine A1 receptor agonist antagonized the development of status (150). Evidence from in vivo microdialysis experiences, performed intraoperatively on humans, suggests that adenosine may be a potential mediator of seizure arrest and postictal refractoriness (151). The clinical use of adenosine and of adenosine analogues has not been investigated extensively (152), and there is evidence that systemically administered adenosine analogues do not cross the blood–brain barrier (153). Moreover, the widespread effects of adenosine in the brain may not permit the development of a highly specific anticonvulsant with minimal CNS side effects.
Catecholamines and Indolamines
Adrenergic neurotransmitters are believed to play a significant role in the regulation of cortical excitability; consequently, a number of laboratories have searched for abnormalities in this system. Brain norepinephrine levels are low in several species of epileptic animals, and a decrease in brain norepinephrine concentration increases their seizure susceptibility. Conversely, an increase in brain norepinephrine levels decreases seizure severity (154). The lesioning of noradrenergic projections from the locus ceruleus lowers the threshold for forebrain and brainstem seizures (155). The importance of the role of brain stem catecholaminergic projections also is suggested by experiments in cats invoving the vagus nerve stimulator (VNS). The efficacy of VNS was lost when the locus coeruleus was lesioned by 6-hydroxydopamine (156).
The serotonergic system also influences the expression of seizures. In view of the recent popularity of highly selective serotonin-uptake antagonists as antidepressants, it is of interest that fluoxetine (Prozac) appears to have anticonvulsant effects (157,158,159). Experiments have suggested that the antiepileptic action of fluoxetine on CA1 neurons is caused by an enhancement of endogenous serotonin that in turn seems mediated by 5-hydroxytryptamine-1A (5-HT1A) receptor (160,161). Interestingly, in a genetic rat model of absence epilepsy, a 5-HT1A agonist increased spike waves in a dose-dependent manner (162). Genetically altered mice lacking the gene for 5-HT2C receptor seem to exhibit spontaneous seizures, while the wild type background could be made to mimic such behavior by the administration of a specific 5-HT2C antagonist (163).
Biochemical Alterations Induced by Seizures
During a brief seizure, the brain undergoes several biochemical alterations. Cerebral oxygen and glucose consumption increase strikingly, but with maintenance of adequate ventilation, the increase in cerebral blood flow is sufficient to meet the increased metabolic requirements of the brain.
These studies, derived from experimental animals, have been confirmed in the epileptic human by PET. Autoradiography using labeled 2-deoxyglucose as a substrate provides excellent information on the local cerebral glucose metabolism. Using this technique, cerebral metabolism is generally found to be increased in the area of the epileptic focus during a seizure. In some instances, the area of increased metabolic activity extends to adjacent tissue and into areas of neuronal projection (164). In absence seizures, a marked and diffuse increase in cerebral metabolic rate occurs (165). The hypermetabolism probably reflects the enhanced excitatory and inhibitory neuronal activity during a seizure.
By contrast, the interictal metabolic picture reveals areas of hypometabolism (128,166). These observations, important to the localization of epileptic foci, are discussed in a subsequent section of this chapter.
When a generalized seizure lasts 30 minutes or longer, it is usually accompanied by apnea. Apnea induces hypoxia and carbon dioxide retention. As a result of the energy demands of the convulsing muscles, the subject becomes hypoxic and hyperpyrexic (167). Oxygen tension within the brain decreases, a shift toward anaerobic metabolism occurs, and lactic acid accumulates (168). MR spectroscopy performed on rabbits subjected to status epilepticus corroborates these data. Phosphocreatine levels decrease, inorganic phosphorus levels increase, lactate increases, and intracellular pH decreases within 30 minutes of the onset of seizures (169). The increase in cerebral lactate after prolonged seizures results from activation of phosphofructokinase, which is a primary regulatory enzyme for cerebral glycolysis (169).
Clinical Manifestations
To facilitate presentation of the clinical manifestations of seizures, which vary even in a given patient, each of the common epilepsies is discussed. A miscellaneous group of less common epilepsies is then covered, with literature references for more extensive reading. Finally, the discussion turns to febrile seizures and the problem of seizures during the neonatal period.
Epilepsies Characterized by Generalized Tonic-Clonic (Grand Mal) Seizures
A generalized tonic-clonic seizure, occurring as a manifestation of primary generalized epilepsy, occurring as a secondary generalization of a partial epilepsy, or alternating with other seizure forms is the most common epileptic manifestation of childhood (see Table 14.3) (170). These conditions do not represent a homogeneous group but are seen in a variety of clinical settings.
The seizure can be primary generalized or secondarily generalized. A primary generalized seizure can occur without warning, whereas a secondarily generalized seizure can be preceded by an aura. Occasionally, the child might be irritable or might manifest unusual behavior for several hours before the seizure. In localizing the epileptic focus, the aura offers the most important clinical clue, more reliable at times than the EEG. The examining physician should always try to elicit its history. The most common epileptic aura is a sensation of dizziness or an unusual feeling of ascending abdominal discomfort. These sensations have been attributed to a discharge in the area of visceral sensory representation but offer less evidence for the site of the epileptic focus than do focal sensory symptoms (171).
In the classic form of an attack, the aura, if present, can be followed by rolling up of the eyes and loss of consciousness. A generalized tonic contraction of the entire body musculature occurs, and the child can utter a piercing, peculiar cry, after which he or she becomes apneic and cyanotic. With the onset of the clonic phase of the convulsion, the trunk and extremities undergo rhythmic contraction and relaxation. As the attack ends, the rate of clonic movements slows, and finally, the movements cease abruptly. The duration of a seizure varies from a few seconds to half an hour or more. In the series of Shinnar and coworkers, new-onset seizures lasted longer than 5 minutes in 50% of children. In 29% of children, they lasted longer than 10 minutes; in 16%, they lasted longer than 20 minutes; and in 12%, they lasted longer than 30 minutes. The authors pointed out that the longer a seizure lasts, the less likely it will stop spontaneously (172). This is commensurate with our experience.
A series of attacks at intervals too brief to allow the child to regain consciousness between attacks is known as status epilepticus. Because status epilepticus is one of the few neurologic conditions requiring emergency treatment, it will be referred to again in the section on treatment.
After the seizure, the child can remain semiconscious and then confused for several hours. When examined soon after an attack, he or she is poorly coordinated, with mild impairment of fine movements. Truncal ataxia, increased deep tendon reflexes, clonus, and extensor plantar responses can be present. Occasionally, the child appears blind and speechless. Postictally, he or she may vomit or complain of severe headache.
The major motor attack has numerous variations. Occasionally, particularly when drug therapy has been partly effective in controlling a secondarily generalized seizure, a typical aura occurs but is not followed by a seizure. In other patients, either the tonic or the clonic phase is too brief to be noted. Attacks can occur at any time of the day or night, although their frequency is somewhat greater shortly before or after the child falls asleep or awakens. Approximately one-fourth of patients experience nocturnal seizures; the remainder experience diurnal or mixed seizures. Generally, patients who for 1 year or more have experienced seizures only during sleep are unlikely to have attacks at other times of the day. In the experience of D’Allessandro and colleagues, who studied both pediatric and adult patients, the risk of a daytime seizure during a six-year follow-up was 13% (173). In some girls, seizures occur a few days before or shortly after their menstrual period.
A generalized tonic-clonic seizure or other epileptic manifestations can be precipitated by infection and fever, fatigue, emotional disturbances, hyperventilation and alkalosis, and drugs.
Fever can induce a seizure not only in children experiencing febrile seizures, but also in patients who previously had recurrent epileptic attacks unassociated with fever. In some children, dehydration and ketosis accompanying an acute infectious illness decrease seizure frequency.
In a few children, excessive fatigue or lack of sleep appears to precede a seizure but probably does not represent an important precipitating factor. Considerable clinical evidence indicates that epileptic children have fewer seizures when engaged in regular strenuous physical activity. Although fatigue seems to have little effect on the EEG, sleep deprivation can activate the EEG of epileptic patients and can precipitate seizures (174).
Parents often believe that emotional disturbances precipitate seizures in epileptic children. No evidence supports this idea, however, and there is no justification for parents’ failing to set limits to the behavior of their epileptic child.
Hyperventilation and alkalosis induce absence attacks in approximately 90% of patients subject to them but are less effective in precipitating other seizure forms.
A variety of drugs can induce single seizures or status epilepticus. In the experience of Messing and coworkers, isoniazid and psychotropic medications accounted for approximately one-half of cases (175). Other drugs implicated in bringing on seizures include cocaine (176), theophylline, and penicillin. Isoniazid (INH) antagonizes glutamic acid decarboxylase by binding with its cofactor, pyridoxal phosphate, thus interfering with the biosynthesis of GABA. Demonstrable decreases in brain GABA levels result from INH administration. An increasing number of patients has presented with status epilepticus and encephalopathy attributable to INH overdoses (176,177). In these cases, immediate administration of pyridoxal (vitamin B6) is crucial. Theophylline acts as an adenosine antagonist, whereas penicillin acts as a GABA antagonist and interferes with benzodiazepine binding in the brain (152). For INH and theophylline, the convulsive effects tend to be dose related (175). Various anticonvulsants, notably phenytoin, when given in toxic amounts,
increase seizure frequency. Trimethadione, which was formerly used in the treatment of absence epilepsy, occasionally precipitated a grand mal seizure within 1 to 2 days of first being administered. The sudden withdrawal of anticonvulsant medication, particularly barbiturates and benzodiazepines, is the most common cause for status epilepticus. Finally, adolescent patients should be warned against excessive alcohol consumption.
increase seizure frequency. Trimethadione, which was formerly used in the treatment of absence epilepsy, occasionally precipitated a grand mal seizure within 1 to 2 days of first being administered. The sudden withdrawal of anticonvulsant medication, particularly barbiturates and benzodiazepines, is the most common cause for status epilepticus. Finally, adolescent patients should be warned against excessive alcohol consumption.
Spontaneous variations in the frequency of attacks are common and must be taken into account when judging the effectiveness of anticonvulsant medication. In the experience of van Donselaar and colleagues, who studied untreated tonic-clonic seizures in children younger than 16 years of age, 42% showed a decelerating pattern, and ultimately became seizure free even in the absence of anticonvulsant treatment. An accelerating pattern of seizure frequency was seen in only 20% of children with four or more untreated seizures (178). Some children have seizures during early childhood, then remain asymptomatic until puberty, when their attacks recur for 4 to 7 years.
Epilepsies with Typical Absence (Petit Mal) Seizures
Typical absence seizures are most commonly identified with absence (petit mal) epilepsy. In its pure form, an absence (petit mal) attack was defined by Gowers as a “transient loss of consciousness without conspicuous convulsions” (48). Absence seizures are relatively uncommon; they comprise between 2 and 11 percent of seizure types in all ages (179).
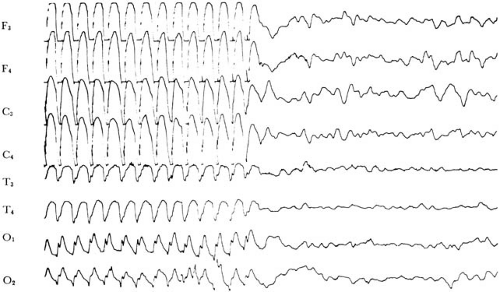 FIGURE 14.3. Three-hertz (three per second) spike-wave discharges in an 11-year-old girl with frequent absence (petit mal) attacks. In this instance, a clinically evident seizure lasting 19 seconds was induced by hyperventilation. (F3, left frontal; F4, right frontal; C3, left central; C4, right central; T3, left temporal; T4, right temporal; O1, left occipital; O2, right occipital.) |
The onset of seizures is usually abrupt, and the child suddenly develops an estimated 20 or more attacks each day. The characteristic attack is a brief arrest of consciousness, usually lasting 5 to 10 seconds, appearing without warning or aura. There can be a slight loss of body tone, causing the child to drop objects from his or her hand, but this loss is rarely profound enough to induce a fall. Minor movements occur in approximately 70% of patients; these are usually lip-smacking or twitching of the eyelids or face, often at the three-per-second frequency of the EEG abnormality (Fig. 14.3). Urinary incontinence is rare (180). The seizure terminates abruptly, and the patient is often unaware of the lapse of consciousness. The occurrence and duration of a lapse can be determined by reciting to the child a series of numbers during the attack and asking the child to repeat them when consciousness appears to have returned. Attacks first appear during childhood; in 64% of instances, they begin between 5 and 9 years of age. They are more common in girls; Dalby’s series had 99 female and 62 male subjects (181).
When attacks are frequent, the child’s intellectual processes are slowed and, often, the first indication of the presence of absence seizures is a deterioration in schoolwork and behavior (182). Responsiveness to auditory stimuli is impaired in the majority of patients in 0.1 to 0.4 seconds after the onset of a generalized spike-wave
paroxysm, but it recovers in 2 to 3 seconds after the cessation of the attack. Extrapolations from spike-wave discharges associated with focal epilepsies have implicated the slow-wave component of the spike-wave complex in the interrupted cognitive function (83). Even brief and clinically undetectable attacks can impair intellectual performance.
paroxysm, but it recovers in 2 to 3 seconds after the cessation of the attack. Extrapolations from spike-wave discharges associated with focal epilepsies have implicated the slow-wave component of the spike-wave complex in the interrupted cognitive function (83). Even brief and clinically undetectable attacks can impair intellectual performance.
Some absence attacks are more complex, and their appearance can be difficult to differentiate from complex partial seizures. They involve brief behavioral automatisms or, less commonly, prolonged symmetric myoclonic movements of the head or extremities (myoclonic petit mal). In the series of Sato and coworkers, some 40% of patients with typical absence seizures also experienced a grand mal attack, either before or after the onset of their absence attack (183). This figure is probably high and perhaps reflects the patterns of referral to a university center.
On a clinical basis, absence attacks must be distinguished from brief complex partial seizures and daydreaming. Complex partial seizures are often preceded by an aura and followed by postictal depression. Additionally, brief complex partial seizures tend to be less frequent and clustered and rarely will occur more than twice a day. Routine EEG studies in most instances clarify the situation. In a child with typical absence attacks, the EEG demonstrates a 3-Hz spike-wave discharge, which occasionally slows as the seizure progresses. The interictal EEG is normal in approximately one-half of patients, and the background EEG frequency is generally age appropriate (184). Approximately 10% of children with clinical and EEG features of typical absence attacks have a focal onset to their EEG seizures and demonstrate focal cortical lesions (185). Attacks of daydreaming also can occur several times a day and can last for several seconds to minutes. Automatisms are absent, there is no postictal depression, and attacks are not induced by hyperventilation or photic stimulation. The EEG is normal.
Typical absence seizures also should be differentiated from atypical absence attacks, which are associated with the Lennox-Gastaut syndrome (atypical petit mal, petit mal variant). This relatively common seizure type is associated with a variety of CNS insults, and the majority of affected children have a significant developmental delay. Unlike typical absence attacks, the frequency of atypical attacks is cyclic, in that seizure-free periods alternate with days or weeks of a high seizure frequency. The EEG shows complexes occurring at 1.5 to 2.5 Hz or multiple spike and wave discharges. Diffuse slowing of background activity was seen in 85% of children in the series of Holmes and coworkers (184).
Absence attacks are a common prelude to juvenile absence epilepsy and start about one to nine years earlier. Its characteristic features consist of typical absence attacks, seen in approximately one-third of patients and commencing later than those of typical childhood absence epilepsy. Brief myoclonic jerks that generally occur on awakening usually develop around 15 years of age (i.e., several years after the appearance of absence attacks), and tonic-clonic seizures follow some months thereafter. Attacks tend to occur much less frequently than they do in absence seizures, usually once a day or less (185). Intelligence is preserved. The interictal EEG in juvenile absence epilepsy demonstrates discharges not only at 3 Hz, but also at 4 to 6 Hz (186,187,188,189). The background activity is usually normal. In the series of Appleton and coworkers, photosensitivity was seen in 90% of patients (190).
Epilepsies with Complex Partial Seizures (Psychomotor Seizures, Temporal Lobe Seizures)
Complex partial seizures have been defined as seizures that arise from a limited area of one cerebral hemisphere and produce a period of impaired consciousness that varies from mild to profound. Although these seizures are most characteristically associated with lesions of the temporal lobe and have been called temporal lobe seizures, they also can be associated with lesions of the frontal (192) or occipital lobes (193). As the terminology indicates, they are focal seizures.
The epilepsies characterized by this seizure type are heterogeneous. Pathologic alterations seen within the surgically resected temporal lobe have been summarized in Table 14.6 (194).
The most common abnormality is MTS (Ammon’s horn sclerosis or hippocampal sclerosis). In the series of Harvey and colleagues, it was seen in 21% of children aged 15 years or younger with new-onset temporal lobe epilepsy (195). The association of this characteristic lesion with both epileptogenicity and seizure-related damage has been discussed in the Neuropathologic Factors section of this chapter. Less commonly, 13% of cases in the series of Harvey and colleagues (195), a variety of tumors in the epileptogenic cortex can occur. These include hamartomas, which occasionally undergo malignant transformation, small gliomatous nodules, hemangiomas, and lesions suggestive of tuberous sclerosis (196,197). In early childhood, however, tumors are the most common etiology for complex partial seizures. Thus, in the series of Wyllie and colleagues, comprising children younger than 12 years of age who underwent temporal lobectomy, tumors were seen in 64%, as compared with MTS, seen in 29%. It is of note that 75% of children with MTS in the series of Wyllie and coworkers had a history of previous febrile convulsions (198). When bilateral MTS develops early in life, usually after prolonged seizures or status epilepticus, the clinical picture is marked by loss of language, or failure of language
development, and impaired social and adaptive learning (199).
development, and impaired social and adaptive learning (199).
In a significant proportion of patients with complex partial seizures, focal abnormalities are found outside the hippocampus, in the limbic portion of the frontal lobes, in the lateral temporal lobe, and in nonlimbic areas outside the temporal lobe (200). In these instances, the ictal discharge probably spreads from the focus to involve the temporal lobe.
Even though seizures start before 10 years of age in approximately 75% of children (95), typical complex partial seizures are rarely seen until 10 years of age. Rather, children who later develop complex partial attacks can have an antecedent history of convulsive seizures associated with chewing, lip-smacking, or other oral automatisms (201). Additionally, they can experience a variety of behavior disorders, enuresis, nightmares, and sleepwalking (202).
Seizure manifestations in older children are presented in Table 14.7. The aura can consist of a variety of subjective phenomena. In children, Glaser and Dixon have found intense anxiety usually associated with visceral sensations to be the most common antecedent to complex partial seizures (203). Wyllie and colleagues have noted that a large proportion of children complain of a “funny” or bad taste (198). An epigastric “rising” sensation also is common. Olfactory hallucinations (uncinate fits) are usually described as unpleasant but unidentifiable odors. Their association with temporal lobe tumors is a matter of some dispute. In Daly’s series, almost 40% of 55 patients who experienced this aura were found to have neoplasms (204). Similarly, in the series of Acharya and colleagues, 73% of patients with such an aura harbored a tumor (205). By contrast, Howe and Gibson found a tumor incidence of only 8.1% in their series of 37 patients, which is comparable with the 9.1% overall incidence of gliomas in patients with temporal lobe seizures (206).
TABLE 14.7 Clinical Manifestation of Complex Partial Seizures in Childhood | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Hallucinatory experiences, most commonly a feeling of déjà vu, an adventitious sense of familiarity, and visual hallucinations, have been reported by children with complex partial seizures (207). According to Mullan and Penfield, they occur more frequently when the focus is in the nondominant temporal lobe (208).
Paroxysmal emotional states, particularly fear, are not rare in children and are commonly reported by parents. Rage reactions or temper tantrums are unusual auras of a complex partial seizure, and purposeful aggressive acts are uncommon in the course of seizure. A detailed history of the aura can assist in localizing the seizure focus, but does not help in lateralizing it. Whereas experiential auras, such as fear and déjà vu, almost invariably originate from the temporal lobes, notably the neocortex (200), cephalic auras, such as a sensation of dizziness or lightheadedness are of less localizing value and can emanate from either frontal or temporal areas. Viscerosensory auras were found to accompany a temporal lobe focus in 76% of subjects, and somatosensory auras accompanied a parieto-occipital focus in 62% (209).
On the basis of the initial seizure manifestations, Escueta and associates have divided complex partial seizures into two types (210). In the more common form, type I, the seizure originates from the temporal lobe. Patients briefly stop all activity after their aura. They stand still, stare, or turn pale. Shortly thereafter, minor motor acts are initiated. Prolonged postictal confusion is common in these patients. In type II, seizures originate from outside the temporal lobe, most commonly from the frontal lobe. In this type, automatisms initiate the attack. Commonly, these involve chewing and smacking movements, purposeless fumbling or patting of the hands, and picking at clothes. Postictal confusion is brief in this group of patients. Drop attacks as part of temporal lobe seizures are unusual in childhood (211).
The final part of the seizure generally involves more complex motor acts. The child might move about the
room, begin to undress, and occasionally utter stereotyped or nonsensical phrases. In the majority of cases, these automatisms usually do not last longer than 5 minutes, although reliable observers have recorded prolonged complex partial seizures. Complex partial status epilepticus (psychomotor status) is extremely rare, and to our knowledge, it has never initiated complex partial epilepsy (212,213). In children, the condition manifests by impaired consciousness with intermittent staring and wandering eye movements and intermittent automatisms, such as picking at clothes. At other times, it can result in a prolonged period of amnesia. The condition should be differentiated from absence status or hysterical amnesia. Depth electrode studies suggest that the seizure focus is more commonly within the frontal lobes (214). A variety of electrocardiographic abnormalities can accompany complex partial seizures. These can be responsible for some of the sudden deaths seen in inadequately treated epileptic patients (215).
room, begin to undress, and occasionally utter stereotyped or nonsensical phrases. In the majority of cases, these automatisms usually do not last longer than 5 minutes, although reliable observers have recorded prolonged complex partial seizures. Complex partial status epilepticus (psychomotor status) is extremely rare, and to our knowledge, it has never initiated complex partial epilepsy (212,213). In children, the condition manifests by impaired consciousness with intermittent staring and wandering eye movements and intermittent automatisms, such as picking at clothes. At other times, it can result in a prolonged period of amnesia. The condition should be differentiated from absence status or hysterical amnesia. Depth electrode studies suggest that the seizure focus is more commonly within the frontal lobes (214). A variety of electrocardiographic abnormalities can accompany complex partial seizures. These can be responsible for some of the sudden deaths seen in inadequately treated epileptic patients (215).
Following the seizure, the patient experiences postictal confusion, drowsiness, or clouding of consciousness. When fully recovered, the child has complete amnesia for the entire attack.
As with major motor seizures, the frequency of complex partial seizures varies, but in contrast to typical absence attacks, more than one to two attacks a day is uncommon (203).
The possibility of complex partial seizures is often raised in a child with behavior problems who has an abnormal EEG result. Aird and Yamamoto examined this question and found that approximately one-half of children with behavior problems have an abnormal EEG result. Of all patients studied, 27% had EEG foci primarily involving the temporal lobe (216). Although some authors vigorously dispute this predilection to behavior disturbances (217), others support it and have found behavior disturbances to be three times as common in this as in the other types of epilepsy (218). Additionally, symptoms suggesting complex partial seizures are highly prevalent among violent juveniles (219). Several further studies have confirmed these observations. In a series published in 1980, the incidence of abnormal behavior was 36% (220). Schoenfeld and coworkers, however, found that children with complex partial seizures are not aggressive or delinquent. Rather they suffer from social withdrawal, various somatic complaints, anxiety, and depression (221). The cause of the seizures, their duration, and the presence of associated grand mal attacks do not seem to affect the likelihood of psychiatric disturbances; however, the frequency of seizures and early seizure onset do (221). Boys whose seizure focus is located in the dominant hemisphere and whose seizures commence between 5 and 10 years of age appear to be particularly vulnerable (218). In most instances, the psychiatric illness commences during adolescence; its manifestation before 12 years of age is unusual.
The basis for these psychiatric phenomena has undergone considerable speculation. The most attractive hypothesis states that recurrent complex partial seizures kindle a limbic dopamine system, whose paroxysmal activity does not produce seizures but does produce serious behavior disturbances (222). The role of limbic kindling in the genesis of psychiatric illness is outside the scope of this text. Adamec and Stark-Adamec and Weiss and Post review this subject (223,224).
Epilepsies Characterized by Simple Partial Seizures (Focal Seizures, Partial Seizures with Elementary Symptoms)
Focal seizures are characterized by the development of localized motor or sensory symptoms without impairment of consciousness. In a large proportion of children, these seizures spread to other parts of the body, ultimately becoming generalized with loss of consciousness. On rare occasions, progression follows an orderly sequence, a phenomenon known as the jacksonian march. This type of a seizure is generally considered to be symptomatic of a structural cortical lesion.
Perhaps the most common focal attack observed in children is the versive seizure (225). These seizures begin in or spread to area 8, the frontal eye field, and the supplementary motor area or the mesial part of the premotor area. They are manifested most often by a turning of the eyes, or the eyes and head, away from the side of the focus. In some patients, the upper extremity on the side toward which the head turns is abducted and extended, and the fingers are clenched. Thus, the child appears to look at his closed fist.
The patient may be aware of this movement or may simultaneously lose consciousness. By means of combined telemetry and EEG monitoring, the cortical areas of discharge responsible for this form of seizure have been found to be either the contralateral temporal lobe or the contralateral frontal lobe, anterior to the rolandic gyrus. Patients whose seizure starts with versive movements and who retain awareness of them tend to have a frontal lobe seizure focus. Patients whose seizure starts with staring and automatisms tend to have a temporal lobe focus (210, 226,227). EEG changes, as recorded on scalp electrodes, are seen in only approximately one-third of patients during telemetry-verified attacks of a variety of simple partial seizures. Therefore, the presence of an unaltered EEG does not speak against the diagnosis (228).
Focal motor seizures are particularly common in hemiplegic children. The epileptic movements are usually clonic and begin in the hemiplegic hand, often heralded by localized sensory symptoms. The clonic movements spread over the entire affected side, ultimately becoming generalized in many cases. Postictal weakness (Todd’s paralysis) commonly follows this kind of seizure and can last for several hours or for a day or more. In the experience
of Kellinghaus and Kotagal, Todd’s paralysis was contralateral to the epileptiform focus in 93% of subjects (229).
of Kellinghaus and Kotagal, Todd’s paralysis was contralateral to the epileptiform focus in 93% of subjects (229).
Whereas adults have a high positive correlation between Todd’s paralysis and a structural cortical lesion, children do not, and often attacks of alternating left- and right-sided focal seizures with postictal paralyses are the initial events in a child with apparently idiopathic epilepsy. This observation probably reflects a smaller and less readily detectable focus in the pediatric population.
A heterogeneous group of conditions are characterized by focal (simple partial) seizures. In approximately one-half of patients, one can document an underlying structural lesion. These lesions include developmental malformations, notably gray matter heterotopias and localized pachygyria, gliosis resulting from asphyxial damage or perinatal and postnatal physical trauma, and a variety of space-occupying lesions. In the remainder of patients with simple partial seizures, no obvious etiology can be demonstrated. Several clinically distinct types of these idiopathic focal epilepsies have been recognized. They are characterized by the absence of anatomic or functional focal lesions and by a benign course with normal intellectual development and spontaneous cure. Aside from febrile seizures and a family history of seizures for approximately one-third of children, no obvious antecedents exist. The frequency of these conditions is difficult to ascertain. Roger and Bureau believe they constitute some 60% of partial epilepsies seen in school-aged children (230).
Rolandic Epilepsy
The most common and most clearly delineated of the idiopathic focal epilepsies is midtemporal epilepsy (sylvian epilepsy, rolandic epilepsy, benign childhood epilepsy with centrotemporal spikes). In the experience of Loiseau and coworkers, it represented 10.7% of children seen in a specialized private practice (231). A high incidence of similar seizures occurs in first-degree relatives, and an autosomal dominant form of rolandic epilepsy has been reported (see Table 14.5) (25,232). In approximately 75% of children, this type of a seizure commences between ages 5 and 10 years. Seizures are infrequent, in the majority of cases occurring less than three or four times a year, and are generally brief. Most characteristically, attacks commence with a somatosensory aura, usually referred to the tongue, cheek, or gums and less often to the abdomen. As a result of motor interference, speech is arrested, the child salivates, and tonic or tonic-clonic movements involve the face. Consciousness is preserved in some 60% of children. Because approximately three-fourths of the attacks occur during sleep, a good patient history is difficult to obtain. The interictal EEG shows midtemporal spikes, probably a reflection of discharges arising from the rolandic cortex (233,234). In about 30% of cases, spikes are only seen during sleep (235). The spikes in this syndrome display a horizontal dipole with maximum negativity in the centrotemporal regions and positivity over the superior frontal area (236). In contrast to other seizures having a temporal spike focus, the prognosis in this condition is excellent (237); most children respond well to anticonvulsant therapy. In the experience of Lombroso, almost 50% were seizure free within 3 years of their first attack, and in more than 30%, the EEG reverted to normal as well (233). Beaussart and Faou share this optimism. In their series of 334 cases, no patient had seizures after age 13 years, even when anticonvulsants had been withdrawn (238).
Benign Epilepsy of Childhood with Occipital Focus (Panayiotopoulos Syndrome)
Benign epilepsy with an occipital focus (239) is somewhat less common, accounting for 6% of all children with seizures (240). The age of onset is variable; ranging between 15 months and 17 years, with the peak age being 5 years (239,240). Seizures are infrequent and tend to mainly occur at night. Attacks during the day are initiated by hemianopia, phosphenes (white or colored luminous spots), or visual hallucinations. Amaurosis or nonvisual symptoms follow. The latter include unilateral convulsions, automatisms, or generalized tonic-clonic seizures. Aphasia is not unusual. Ictal deviation and vomiting may be seen, and in about one-half of children, there is postictal migraine (241). The interictal EEG most commonly demonstrates nearly continuous unilateral or bilateral high-voltage spike-wave discharges from the occipital or posterior temporal regions. This is generally suppressed by opening of the eye. The prognosis is excellent with or without anticonvulsant therapy, and all children in Beaumanoir’s series were seizure free with a normal EEG by 9 to 13 years of age (239). The experience of Panayiotopoulos is similar, with 89% of children gaining remission (242).
Focal Epilepsy with Midtemporal Spikes and Affective Symptoms
European epileptologists also distinguish a focal epilepsy with midtemporal spikes and affective symptoms. Seizures commence with a sensation of fear or terror and proceed with chewing and swallowing movements and speech arrest. Consciousness is depressed. Unlike complex partial seizures, seizures cease with therapy, and no evidence for a focal lesion can be found on clinical examination or by imaging studies (228).
Lennox-Gastaut Syndrome (Myoclonic-Astatic Petit Mal)
The epileptic syndromes considered in this section share an early onset, an EEG picture characterized by slow spike-wave forms at a frequency lower than the regular 3 Hz, and
poor prognosis, in terms of both seizure control and ultimate intellectual development. Livingston and associates have used the term minor motor seizures to designate the epilepsies described in this section (243). This term has been abandoned in recent years; not only is there nothing minor about these seizures, but the term is overly broad and encompasses a variety of seizures with limited expression, regardless of their etiology and EEG features.
poor prognosis, in terms of both seizure control and ultimate intellectual development. Livingston and associates have used the term minor motor seizures to designate the epilepsies described in this section (243). This term has been abandoned in recent years; not only is there nothing minor about these seizures, but the term is overly broad and encompasses a variety of seizures with limited expression, regardless of their etiology and EEG features.
The causes for Lennox-Gastaut syndrome (LGS) are multiple. In the experience of Lennox and Davis, compiled in 1950, 50% of patients had a history of perinatal cerebral injury, another 20% might have had an attack of encephalitis or meningitis, and 13% had major complications of gestation (244). Currently, complications of gestation appear to be the most significant cause, with perinatal asphyxia, infections, and genetic factors following in order of importance.
Types of Seizures
Based on the different clinical manifestations of the epileptic attacks, at least four seizure types can be distinguished: atonic (astatic or akinetic) seizures, brief tonic seizures, atypical petit mal seizures, and myoclonic seizures (245).
Atonic (Akinetic) Seizures.
Atonic seizures are characterized by a sudden, momentary loss of posture or muscle tone. In the infant who is able to sit but cannot yet stand, atonic spells consist of a sudden dropping forward of the head and neck, the salaam seizure, such as is seen as part of infantile spasms. In older children, the loss of postural tone precipitates the child violently to the ground. Consciousness is lost only momentarily, but the force of the fall commonly produces injuries to the face and head. Atonic spells recur frequently during the course of a day and are particularly common during the morning hours and shortly after the child awakens.
Tonic Seizures.
Tonic seizures are characterized by a brief generalized increase in muscle tone. These seizures are frequent during non–rapid eye movement (NREM) sleep and generally are the most common form of seizure in LGS as well as the type most resistant to anticonvulsant therapy (246).
Atypical Petit Mal Seizures.
Atypical petit mal seizures are characterized by absence seizures that, unlike true petit mal, tend to occur in cycles and can disappear for periods of several days (247). These seizures can occur by themselves or can be accompanied by grand mal or complex partial seizures.
Myoclonic Seizures.
The term myoclonic seizure includes a variety of seizures characterized by myoclonus, that is, single or repetitive contractures of a muscle or a group of muscles. For therapeutic and prognostic purposes, myoclonus has been classified into two forms: epileptic and nonepileptic (248). Myoclonic seizures account for approximately 7% of the epilepsies that have an onset during the first 3 years of life (249). In primary generalized epileptic myoclonus, the myoclonic jerks consist of small, random, recurring twitches, most evident in the fingers and hands. They synchronously involve muscles on both sides of the body. An EEG abnormality, originating most commonly from the frontal leads, precedes the myoclonus, suggesting that a hyperexcitable cortex responds to a subcortical input with the paroxysm. This form of myoclonus usually is seen with chronic epileptic disorders. In the series of Wilkins and associates, it was part of LGS in more than one-half of the subjects (250).
Onset of LGS seizures ranges from age 6 months to approximately age 16 years. In some two-thirds of instances, the onset is between 2 and 14 years of age (251). In some patients, 6% of Kruse’s series, LGS follows infantile spasms (251). In approximately one-half of the children, one or more major motor attacks, with or without fever, precede the illness. Brett delineated a group of children who experience a sudden onset of major seizures that, after a seizure-free interval of approximately 1 week, are followed by LGS and progressive intellectual deterioration (252). This entity, termed epileptogenic encephalopathy by Brett, is not rare. Its cause is unknown, but in view of the occasional cerebrospinal fluid (CSF) pleocytosis, it might be infectious. The association of Ohtahara syndrome or early infantile epileptic encephalopathy (EIEE) to West syndrome and LGS has been documented (253). Atonic, myoclonic, or atypical absence seizures can continue throughout the patient’s lifetime. In three-fourths of patients, generalized tonic-clonic or focal seizures also can appear.
LGS syndrome correlates with an EEG result that Lennox and Davis termed the petit mal variant (Fig. 14.4) (244). It is an asymmetric, sometimes lateralized, slow (2.0 to 2.5 Hz) polyspike-wave discharge (atypical spike-wave discharge), which, in contrast to the 3-Hz synchronous discharge of petit mal, is less likely to be provoked by hyperventilation.
Mental development of the child with LGS is usually slow. In part, this reflects the underlying brain disease, and in part, it is the result of the frequency of seizures. Characteristically, the earlier motor milestones are attained at the expected age, but subsequent evaluations reveal subnormal intelligence in a large proportion of affected children. In the series of Blume and associates, 35% of patients attained an IQ of 75 or above, and only 10% scored above 100 on psychometric testing (254). In the experience of Chevrie and Aicardi, the incidence of retardation was even higher (224).
 FIGURE 14.4. Atypical polyspike-wave discharge. Photically (5 Hz) induced myoclonic seizure in a 14-year-old girl. The electroencephalogram shows 3-Hz multiple spike-wave discharges. (F8T4, right frontotemporal; T3T5, left frontotemporal; T4T6, right temporal; FP1C3, left frontopolar-central; FP2C4, right frontopolar-central; C3O1, left central-occipital; C4O2, right central-occipital. Top lead is photostimulator.) |
Differential Diagnosis of Myoclonic Seizures
Pseudo-Lennox syndrome is a condition that was first described by Aicardi and Chevrie under the term atypical benign partial epilepsy of childhood. It is marked by generalized minor seizures, such as atonic seizures, atypical absences, and myoclonic seizures, and focal sharp slow waves and spikes with generalization during sleep. Seizure onset peaked at about three years of age (255). In contrast to LGS, prognosis in terms of seizure remission is good. In the series of Hahn and coworkers, 84% of children with this condition ultimately went into remission (256). However, over 50% of children were attending schools for the mentally handicapped.
Myoclonic seizures also are seen as a nonspecific symptom in several forms of viral encephalitis, metabolic disturbances such as uremia, and progressive cerebral degenerative diseases such as the various lysosomal storage diseases, some of the leukodystrophies, Menkes disease, and Unverricht-Lundberg disease (see Chapter 3).
Several types of nonepileptic myoclonus have been recognized. Cortical reflex myoclonus is believed to result from hyperexcitability of a small region of the sensory portion of the sensorimotor cortex. The muscular contractions are irregular and are precipitated or aggravated by sensory stimuli, most commonly by light, noise, or tapping on the face or chest. Postural changes intensify the contractions. The movements disappear in sleep. The EEG is clearly abnormal, and a paroxysm precedes both the spontaneous and the reflex-induced myoclonic jerks. Giant somatosensory-evoked potentials are characteristic for this type of myoclonus (257).
In reticular reflex myoclonus, the myoclonic jerks affect the entire body, with the flexor muscles being more involved than the extensors. The seizures are believed to be the result of hyperexcitability of the caudal brainstem reticular formation, possibly the nucleus reticularis gigantocellularis (248). An EEG spike usually follows the first electromyographic evidence of myoclonus. Another nonepileptic form of myoclonus is considered to be of spinal cord or brainstem origin. The muscular contractions are rhythmic and are unaffected by sensory stimuli. They persist during sleep. The EEG can be normal or abnormal, and response to anticonvulsant medication is poor.
Nonepileptic myoclonus also is encountered in the context of involuntary movements in some of the disorders of the extrapyramidal system. It is seen in normal individuals while falling asleep. Rarely, frequent but nonprogressive myoclonic movements occur unaccompanied by other types of epileptic attacks or intellectual subnormality. This condition is termed paramyoclonus multiplex or essential myoclonus. It is probably identical to myoclonic dystonia (DYT 11) and is considered with the various dystonias covered in Chapter 3 (258). Closely related to this condition is the exaggerated startle response (hyperekplexia), conditions also considered in Chapter 3. In hyperekplexia, patients exhibit momentary muscular stiffness and loss of voluntary postural control without loss of consciousness.
Other features of this condition include transient hypertonia during infancy, nocturnal jerking of the legs, and insecure gait. No other epileptic phenomena occur, although the EEG can have paroxysmal features. Minor, quantitatively less severe forms of this condition also have been observed. The condition should be distinguished from startle epilepsy, a type of reflex epilepsy (259).
Other features of this condition include transient hypertonia during infancy, nocturnal jerking of the legs, and insecure gait. No other epileptic phenomena occur, although the EEG can have paroxysmal features. Minor, quantitatively less severe forms of this condition also have been observed. The condition should be distinguished from startle epilepsy, a type of reflex epilepsy (259).
Infantile Spasms (West Syndrome)
Infantile spasms most commonly develop between 3 and 8 months of age, with only 8% of cases first being encountered in infants older than 2 years of age. It occurs somewhat more frequently in male infants. Attacks are characterized by a series of sudden muscular contractions by which the head is flexed, the arms are extended, and the legs are drawn up. A cry or giggling can precede or follow the seizure, and the infant can flush or can turn pale or cyanotic.
Other clinical presentations of infantile spasms occur less commonly. They include head nodding and extensor spasms characterized by extension rather than flexion of arms, legs, and trunk. Rarely, the attacks are concluded by a brief clonic seizure.
Lightning attacks (Blitzkrämpfe) (260) are a variant involving a single, momentary, shocklike contraction of the entire body (261).
Clusters of seizures recur frequently, particularly on waking, and some children have 50 to 100 each day. In Jeavons and Bower’s series of 112 children, mental development was normal up to the onset of seizures in 52% and was definitely or probably delayed in the remainder (262). More recent series have had a higher incidence of delayed mental development before the onset of seizures. In 66% of patients, the EEG has the characteristics of hypsarrhythmia, namely diffuse dysrhythmia with high-voltage slow waves and multiple spike-wave discharges (Fig. 14.5) (262). This unique electrical pattern is seen in the early stages of the disorder, becoming apparent after 3 to 4 months of age. In some infants, it is most evident in non-REM sleep. During REM sleep and immediately after arousal from REM or non-REM sleep, the EEG can be normal for up to several minutes (263). The discharges tend to favor the posterior areas of the brain. This contrasts with the paroxysmal discharges in LGS, which are most evident anteriorly. The significance of this finding is not clear, particularly because many children with infantile spasms progress to LGS. To our knowledge, there has not been a systematic study of EEG maturation in infantile spasms. Variations in this pattern are common (modified hypsarrhythmia). They include hypsarrhythmia with a focus of abnormal discharge, hypsarrhythmia with increased interhemispheric synchronization, hypsarrhythmia with little spike or sharp-wave activity, and hypsarrhythmia with episodes of voltage attenuation, a variant termed suppression burst variant by Hrachovy and coworkers (263).
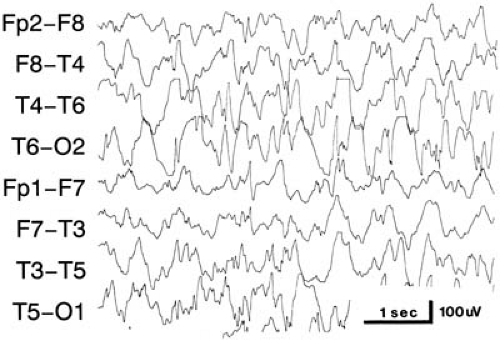 FIGURE 14.5. Hypsarrhythmia in a 6-month-old girl with infantile spasms. The record is characterized by mountainous slow waves, multifocal spikes, and sharp waves. (Fp2–F8, right frontopolar-frontal; F8–T4, right frontotemporal; T4–T6, right temporal; T6–O2, right temporal-occipital; Fp1–F7, left frontopolar-frontal; F7–T3, left frontotemporal; T3–T5, left temporal; T5–O1, left temporal-occipital.) |
Hypsarrhythmia persists for some years, then is superseded by a variety of other paroxysmal abnormalities. These include focal discharges, most commonly arising from the temporal areas, multifocal spike discharges, and an atypical spike-wave pattern. The evolution of infantile spasms to LGS was commented upon earlier in the discussion of the latter syndrome.
Attacks of spasms are often associated with a discharge of multiple spike-wave or polyspikes. Sudden suppression of electric activity begins with an attack and persists briefly after its completion.
Although infantile spasms are considered as a generalized epilepsy, the neuroanatomic substrates responsible for them are poorly understood (264). A marked decrease in REM sleep has led to the proposal that the basic abnormality is at the pontile level in proximity to centers regulating sleep cycles (265). This hypothesis is supported by the finding on interictal PET scan of hypermetabolism of the brainstem. Additionally, hypermetabolism of the lenticular nucleus occurs, which suggested to Chugani and coworkers a neuronal circuitry involving cortex, lenticular nucleus, and brainstem in the generation of infantile spasms (266).
Infantile spasms are classified as cryptogenic or symptomatic. Cryptogenic spasms, a minority of the cases of infantile spasms [less than 15% in the series of Riikonen (267)], occur in infants with normal birth and development until the onset of seizures, in whom no obvious cause for the convulsions can be demonstrated. A variety of prenatal and perinatal insults are responsible for the majority
of cases in the symptomatic group. Abnormalities of gestation, manifested by a history of maternal infection and prematurity, are particularly common. Perinatal asphyxia or traumatic birth injuries are seen less often. Neonatal seizures are a common precursor, and symptomatic neonatal hypoglycemia is encountered in some 18% of infants (267). In the remainder of the infants, tuberous sclerosis is the major etiologic factor, with intracerebral calcifications detectable by CT scanning in up to 25%. Infantile spasms also are seen in neurofibromatosis, agenesis of the corpus callosum, and metabolic diseases such as phenylketonuria, maple syrup urine disease, and pyridoxine dependency (268). In a small group of infants, spasms begin shortly after pertussis immunization with the whole cell vaccine. Whether this association is fortuitous, as suggested by Bellman and coworkers (269), or whether pertussis vaccine indeed represents a rare cause for infantile spasms will probably never be resolved, and with the switch to acellular pertussis vaccine, this question has become moot (270).
of cases in the symptomatic group. Abnormalities of gestation, manifested by a history of maternal infection and prematurity, are particularly common. Perinatal asphyxia or traumatic birth injuries are seen less often. Neonatal seizures are a common precursor, and symptomatic neonatal hypoglycemia is encountered in some 18% of infants (267). In the remainder of the infants, tuberous sclerosis is the major etiologic factor, with intracerebral calcifications detectable by CT scanning in up to 25%. Infantile spasms also are seen in neurofibromatosis, agenesis of the corpus callosum, and metabolic diseases such as phenylketonuria, maple syrup urine disease, and pyridoxine dependency (268). In a small group of infants, spasms begin shortly after pertussis immunization with the whole cell vaccine. Whether this association is fortuitous, as suggested by Bellman and coworkers (269), or whether pertussis vaccine indeed represents a rare cause for infantile spasms will probably never be resolved, and with the switch to acellular pertussis vaccine, this question has become moot (270).
As might be expected, a variety of neuropathologic findings have been reported. These include structural malformations of cortical gray matter. The most commonly observed abnormality observed in surgically resected specimens is a focal cortical dysplasia (271). Other alterations include hamartomatous proliferation of multipotential neuroectodermal cells (272), ulegyria, lissencephaly, unilateral megalencephaly, and pachygyria (273). Focal areas of cortical microdysgenesis are not readily detectable by magnetic resonance imaging (MRI) during the first few months of life but can be visualized indirectly in subsequent studies by a focal delay in maturation of subcortical white matter. This can only be visualized by repeated imaging studies (274). Tumors, cytomegalovirus infection (275) and spongy degeneration of gray and white matter are less common (271,276).
In most children, the outlook for normal intellectual development is poor, even though in approximately 50%, infantile spasms have ceased by 3 years of age or are replaced by the LGS or by major motor attacks (277). According to Jeavons and Bower, none of the patients whose development was already retarded when infantile spasms began had a subsequent normal intellectual development, whereas 29% of infants believed to have developed normally up to the onset of seizures (cryptogenic infantile spasms) were found to be neurologically intact and intellectually in the normal or low-normal range when their infantile spasms disappeared (262). More recent series, such as those of Riikonen (267) and Gaily and coworkers (278), present comparable results. The latter workers observed 67% of infants with cryptogenic infantile spasms to have normal intelligence when tested between the ages of 4 and 6 years. Specific cognitive deficits were found in 42% of children in this group. In the series of Kivity and coworkers, the outcome of children with cryptogenic infantile spasms treated with high doses of adrenocorticotropic hormone prior to mental deterioration was uniformly excellent (279). Riikonen found that in children whose infantile spasms are attributed to tuberose sclerosis, the long-term prognosis is worse than in other symptomatic or cryptogenic cases of infantile spasms (280). By contrast, the prognosis is relatively good when infantile spasms accompany neurofibromatosis (281).
Differential Diagnosis
A condition that has a similar ominous prognosis as infantile spasms, and also is marked by frequent minor generalized seizures is Ohtahara syndrome (282) Onset of seizures is earlier than in infantile spasms and is usually prior to three months. The main seizure type is tonic spasms with or without clustering. The EEG is characteristic. It consists of high voltage bursts interspersed with a nearly flat pattern. The condition results from a variety of brain malformations, notably focal cortical dysplasias. It is more resistant to treatment than infantile spasms, and the prognosis for mental function is poor. In many instances, the tonic spasms evolve into infantile spasms, and like infantile spasms into LGS (283).
Treatment
The relationship between treatment regimens and outcome is controversial, and the recommendations prepared by the American Academy of Pediatrics and the Child Neurology Society are too conservative to clarify the situation. These bodies concluded that adrenocorticotropic hormone (ACTH) is “probably an effective agent” for the short-time treatment of infantile spasms. They also concluded that there was insufficient evidence to state that successful treatment of infantile spasms improves the long-term prognosis (284). The Israeli group under Lerman and Kivity, however, found that optimal results are obtained when treatment is initiated within a month after the onset of seizures and when high doses are given for a prolonged period (259,285), and as already noted, their results with early treatment of infants with cryptogenic infantile spasms were uniformly excellent in terms of mental development and seizure control. The series of Glaze and coworkers did not have a sufficient number of cryptogenic cases to determine the benefit of early therapy (286). The more recent series of Askalan and coworkers also is too small and included infants who already had shown developmental regression before treatment (286a). Certainly, early ACTH therapy does not alter the uniformly poor outcome of symptomatic infantile spasms (286).
In this respect, it is of interest that the relapse after ACTH treatment is much higher in patients with tuberose sclerosis than in other forms of symptomatic infantile spasms (280). Even when treatment does not improve mental functioning, it can have a beneficial effect on seizure control and on the EEG in over one-half of the cases.
In this respect, it is of interest that the relapse after ACTH treatment is much higher in patients with tuberose sclerosis than in other forms of symptomatic infantile spasms (280). Even when treatment does not improve mental functioning, it can have a beneficial effect on seizure control and on the EEG in over one-half of the cases.
Many regimens for ACTH treatment have been proposed. As a rule, at higher ACTH doses, there is a greater proportion of remissions but also a higher incidence of major adverse reactions. One convenient treatment schedule follows: ACTH is given intramuscularly in a daily dose of 80 to 120 U. This dose is maintained until spasms and hypsarrhythmia disappear. If a response is noted, the dose is changed to 40 to 80 U every other day and then gradually reduced to half that dose. This is maintained for approximately 3 more months. Thereafter, the dosage is reduced further by 10 U at 2-month intervals until the drug is completely withdrawn or the minimal dosage for seizure control is reached. One of us (JHM) has recommended a starting dose of 40 U, which is increased at weekly increments to 60 U and 80 U if there has not been any response. A prospective study has indicated that there is no difference in the effectiveness of high-dose and low-dose regimens with respect to seizure control and normalization of the EEG (287). Ito and coworkers have suggested a starting dose of 20 U of synthetic ACTH for infants over 1 year of age and 10 U for infants under 1 year of age (288). On this dosage, excellent seizure control was seen in 76% of patients, and normalization of EEG was seen in 38%. Outcome in terms of mental function was poor, however, with only 6% of subjects being normal.
Side reactions to ACTH include hypertension, which can become apparent at a dosage of 80 U per day, gastroenteritis, sepsis, osteoporosis, and unexplained CNS hemorrhage. The incidence of these complications varies considerably and probably reflects the different treatment schedules. In some series, it can be as high as 37% (289), whereas others have not shown any serious morbidity (290). One interesting concomitant to ACTH therapy is an enlargement of the cerebral ventricular system, which is disclosed by serial neuroimaging studies. This enlargement, which is occasionally permanent (perhaps as a result of abnormalities predating the onset of infantile spasms), is believed to reflect loss of water, owing to altered CSF absorption (291).
The mechanism by which ACTH controls infantile spasms is a matter of some debate, and Snead has reviewed the various hypotheses (292). Baram and colleagues have shown that in infantile spasms, CSF levels of ACTH and cortisol are reduced and have postulated that there is an increased synthesis of corticotropin-releasing hormone (CRH), which in infant rats has been shown to induce seizures originating from the amygdala. They suggest that ACTH desensitizes the CRH receptors and thus induces a negative feedback on CRH release (293). ACTH also could modulate second messenger systems or increase the expression of several genes and thus accelerate myelination, in this manner shortening a vulnerable, hyperexcitable period (294). In that respect, it is significant that several studies, as well as our own experience, indicate that ACTH is a potent anticonvulsant for Lennox-Gastaut syndrome (290,295).
Of the other medications recommended for the treatment of infantile spasms, vigabatrin appears to be the most effective. In an uncontrolled study, Mitchell and Shah started patients on 125 to 250 mg per day, with gradual increments to a target dose of 100 mg/kg per day or any lower effective dose (296). In this unselected series, 60% responded with cessation of spasms and resolution of hypsarrhythmia. Appleton recommends starting at 25 to 50 mg/kg per day and increasing the dosage to a maximum of 80 to 120 mg/kg per day. In their group of children, making up 81% of symptomatic infantile spasms, 81% experienced total cessation of spasms. On follow-up 2 years later, 67% had remained seizure free (297). Chiron and coworkers have used vigabatrin with considerable success in the treatment of infantile spasms associated with tuberose sclerosis (298). In a retrospective study by Cossette and colleagues, vigabatrin was found to be as effective as ACTH in terms of controlling seizures and offering a good developmental outcome. Side effects were less frequent in vigabatrin-treated children than in those who received ACTH (299). Their results are at variance with those of Lux and colleagues, who found that synthetic adrenocorticotropic hormone and prednisolone were more likely to induce cessation of spasms than vigabatrin given in minimal doses of 100 mg/kg per day (299a). The high incidence of visual field defects seen on even low doses of vigabatrin has raised concerns with respect to the safety of the medication. On that account, Riikonen believes that the benefits of vigabatrin do not outweigh the risks of irreversible visual changes (300). In spite of these drawbacks, vigabatrin has become the first-line drug in other clinics for the treatment of infantile spasms, particularly when these occur in a setting of tuberose sclerosis (301,302).
Oral steroids are probably not as effective for infantile spasms as ACTH. In a prospective, randomized study, high-dose ACTH (150 U/m2 per day) was more effective than prednisone by both clinical and EEG criteria (303). Prats and coworkers have suggested treating infantile spasms with extremely high doses of sodium valproate (100 to 300 mg/kg per day). According to their protocol, the anticonvulsant is given for 21 days or until the EEG resolves. Then, the medication is reduced to 25 to 50 mg/kg per day (304). Although their results require confirmation, the outcome of children treated with valproate appears to be as good or better than those treated with ACTH both in terms of seizure control and normal intellectual development.
Kossoff and coworkers have used the ketogenic diet to treat infantile spasms and found the response rate to be significant, with 46% being more than 90% improved and
100% of subjects being more than 50% improved in terms of EEGs and parent reports (305).
100% of subjects being more than 50% improved in terms of EEGs and parent reports (305).
Many other treatment regimens have been suggested. In Japan, large doses of pyridoxine are preferred by the majority of institutions, followed by the combination of pyridoxine and valproate, and valproate monotherapy (306). Combined treatment with vigabatrin and topiramate has been used in Italy (307). Zonisamide has been found to be effective in one-fourth to one-third of patients with infantile spasms (308,309,310).
As ominous as are infantile spasms in terms of ultimate mental development, Lombroso and Fejerman were able to distinguish a benign form (311). In this condition, myoclonic attacks are accompanied by normal intellectual development and a normal EEG, in contrast to infantile spasms in which the EEG is abnormal at the onset of seizures or becomes abnormal within a few months, as is the case in young infants. In the benign form of the condition, myoclonus ceases before 2 years of age, and no other seizures supervene.
Attacks characterized by shuddering and resembling myoclonic seizures can occur in monosodium glutamate poisoning and also can be noted as a prelude to familial (essential) tremor (see Chapter 3).
Juvenile Absence Epilepsy; Juvenile Myoclonic Epilepsy
Juvenile absence epilepsy and Juvenile myoclonic epilepsy (JME) are two clinically related conditions that are genetically heterogeneous, probably transmitted as a dominant trait. Several genes have been implicated in this condition. One of them encodes the GABA receptor (GABABR1) and has been mapped to chromosome 6p21.3 (Table 14.5) (315,316). Another gene (EFHC1) has been mapped to chromosome 6p12–p11 and encodes a protein with an EF-hand motif. EF hands are Ca(2+) binding motifs that are widely distributed throughout the entire animal kingdom (317).
Juvenile myoclonic epilepsy has a prevalence of 0.5 to 1.0 per 1,000 and represents some 4% of the primary generalized epilepsies (189).
Symptoms usually become apparent in adolescence, with age of onset between 12 and 18 years (189). Seizures are characterized by sudden myoclonic jerks of shoulders and arms that usually appear shortly after awakening. A majority of patients also experience grand mal attacks. As a rule, myoclonic jerks precede grand mal seizures. Up to one-third of patients also experience absence attacks, which tend to precede the onset of myoclonic jerks (189). Intelligence remains normal. The interictal EEG is characteristic, in that it demonstrates 4- to 6-Hz polyspike and wave complexes, with photosensitivity in approximately 30% (318). Valproic acid is effective in up to 90% of subjects but has to be continued for the remainder of the patient’s life because withdrawal of anticonvulsants results in seizure recurrence in some 90% of patients, even after many years of complete control (319). Alternatives to valproate therapy with newer anticonvulsant medications are being explored and are reviewed by one of us (R.S.) (320).
Miscellaneous Seizure Forms
A number of epileptic conditions are manifested by unusual seizure forms. These are seen too rarely to warrant more than a brief description. The following types deserve mention.
Abdominal Seizures
Paroxysmal attacks of abdominal pain can occur as an aura for a major motor attack or can be the only manifestation of a seizure. The abdominal pain is usually periumbilical, radiating to the epigastrium. In the majority of cases, it lasts 5 to 10 minutes, but it can persist for 24 to 36 hours. It is usually associated with disturbed awareness (321).
The pediatrician often sees a child who has recurrent bouts of paroxysmal abdominal pain accompanied by vomiting and in whom the usual gastrointestinal evaluation result has been normal. Only a small proportion of these children has abdominal epilepsy. A more common cause of this complaint is childhood migraine. A history of pain and vomiting is common in small children who subsequently develop the usual clinical picture of migraine. The diagnosis of abdominal epilepsy rests on the presence of other epileptic manifestations, usually complex partial seizures, an abnormal EEG pattern during or between attacks, and, less convincingly, a favorable response to anticonvulsants, usually carbamazepine, valproate, or phenytoin (322).
Epilepsia Partialis Continua
Epilepsia partialis continua, a variant of a simple partial (focal) seizure, is characterized by clonic movements, usually localized to the face or upper extremities, that persist over long periods either continuously or with only brief interruptions. Consciousness is not impaired, but postictal weakness is usually evident. We have seen several children with this condition who had an underlying chronic encephalitis of the kind described by Aguilar and Rasmussen (61) and Rasmussen and McCann (323). Rasmussen syndrome was the most common cause for epilepsia partialis continua in the series of Thomas and coworkers (324). This condition, and its relationship to immune disorders and a
variety of infectious agents, and its treatment is considered in Chapter 8.
variety of infectious agents, and its treatment is considered in Chapter 8.
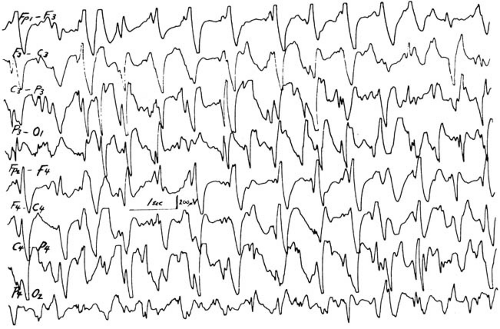 FIGURE 14.6. Absence status (generalized nonconvulsive status) in a 26-month-old girl with mild retardation and 14-day history of drowsiness. Note the enormously high voltage of the spike and sharp waves. (C, central; F, frontal; O, occipital; P, parietal.) (Marker indicates 200 μV.) (Courtesy of Dr. E. Niedermeyer, Johns Hopkins Hospital, Baltimore, MD.) |
Various space-occupying lesions, including tumor, abscess, and cerebral cysticercosis, are less frequent causes for epilepsia partialis continua. Epilepsia partialis continua also has been encountered in various metabolic disorders, notably nonketotic hyperglycinemia (see Chapter 1).
Nonconvulsive Status Epilepticus, Absence Status (Generalized Nonconvulsive Status Epilepticus, Petit Mal Status)
Several types of nonconvulsive status epilepticus (NCSE) have been recognized: a generalized form, also termed absence status or petit mal status, and a partial type, termed complex partial status epilepticus; NCSE in patients with learning difficulties—including status epilepticus during sleep, atypical absence status epilepticus, and tonic status epilepticus—and NCSE in coma (325,326). NCSE is frequently seen in patients who are critically ill, such as in end-stage renal disease, or who have other major medical problems and can arise from treatment with cephalosporins, cefepine, isfosfamide, or tiagabine or upon acute withdrawal of diazepines, such as lorazepam.
Absence status is the most common form of nonconvulsive status epilepticus. It is characterized by a prolonged state of clouded mental activity usually accompanied by an EEG picture of atypical, slow spike-wave complexes and polyspike discharges. Untreated, the condition can last from several hours to as long as 2 years (327). Absence status almost always occurs in children with pre-existing organic cerebral lesions, a history of intellectual retardation, and a variety of other seizure forms (Fig. 14.6) (328). Approximately one-fourth of children with Lennox-Gastaut syndrome enter absence status one or more times during their lifetime (251).
Farran and coworkers have suggested that absence status is seen in patients with cerebellar anomalies and a consequent defect in the inhibitory system mediated by the Purkinje cells (329). The PET scan obtained during an attack fails to confirm this supposition in that it demonstrates only diffuse hypometabolism of fluorodeoxyglucose (330).
Complex partial status epilepticus is characterized by altered mentation, with impaired or absent language function, and a variable degree of responsiveness. The EEG during an attack shows various abnormalities, ranging from focal temporal spike and wave discharges to rhythmic slowing.
Benzodiazepines (lorazepam or diazepam) or valproate are the best drugs for treatment of both forms of nonconvulsive status, followed by institution or resumption
of long-term anticonvulsant therapy (331). There is no conclusive evidence that prolonged NCSE can induce brain damage, and Kaplan cautions against overtreatment of the condition (332).
of long-term anticonvulsant therapy (331). There is no conclusive evidence that prolonged NCSE can induce brain damage, and Kaplan cautions against overtreatment of the condition (332).
Landau-Kleffner Syndrome
Landau-Kleffner syndrome, which has attracted considerable attention in recent years, is marked by an acquired aphasia in children who have had normal language and motor development (333). Symptoms develop between 4 and 7 years of age. A variety of seizures accompany this condition in approximately 80% of cases. They can be rare and can disappear completely before adolescence. The EEG abnormalities are diagnostic. They include bilateral independent temporal or temporoparietal spikes, or spike and wave discharges, and in a large proportion of children, a continuous spike and wave discharge during as much as 90% of the sleeping state (334). Neuroimaging study results are normal. The aphasia frequently fluctuates in severity, but as a rule, the younger the age when symptoms start, the worse the prognosis in terms of language function (335). Other aspects of higher cortical function are preserved. Although in the initial report the condition was seen in two brothers, the etiology remains totally unknown.
Valproate, ethosuximide, and the benzodiazepines can improve the condition; phenobarbital and carbamazepine are generally ineffective. ACTH or corticosteroids also can be partially effective (336). Other therapeutic approaches include the use of intravenous immunoglobulins, which can be effective in some patients (337), and multiple subpial transections (338). As a rule, the EEG abnormalities regress with time, leaving the child with a severe receptive and expressive aphasia.
Landau-Kleffner syndrome has much in common with a condition in which subclinical electrical status epilepticus occurs during slow-wave sleep; continuous spike-wave discharges during slow-wave sleep (ESES) (339,340). This is a relatively rare seizure disorder. It is marked by partial or generalized seizures that are usually nocturnal, associated with reduction in language function, reduced attention span, and a variety of behavior disorders (341,342). The characteristic EEG pattern appears with onset of sleep and consists of spike-wave discharges, usually at 1.5 to 2 Hz, that persist throughout slow-wave sleep. During REM sleep, the frequency of spike and wave discharges is the same as during the waking state.
TABLE 14.8 Unusual Seizure Forms | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Treatment of the EEG abnormalities is not successful, although a number of anticonvulsants and corticosteroids have been used (341). In distinction from Landau-Kleffner syndrome, where there is an isolated disturbance of language function, other cognitive functions also are affected in ESES. The clinical course and response to anticonvulsants is similar for the two conditions, and many workers consider ESES to be a variant of the Landau-Kleffner syndrome or to represent a clinical continuum (334,340).
Other Seizures
Rarer seizure forms are summarized in Table 14.8 (343,344,345,346,347,348,349,350,351). The most likely of these to be encountered are the reflex seizures. This term denotes seizures that are repeatedly initiated by clearly defined stimuli. Reflex epilepsies account for some 5% of all epilepsies, with visually induced reflex (photogenic) epilepsy being the most common form, constituting 53% of Forster’s series (352). Visual reflex epilepsy includes television epilepsy and video game epilepsy as well as most cases of epilepsy in which the child induces his or her own seizures by looking at a light source and rapidly moving the hand back and forth in front of the eyes (“fanning”) (353,354). Other patients, not included in the group of reflex epileptic patients, can have both photogenic and nonphotogenic seizures or, even more
commonly, can show only a photoconvulsive response on the EEG (355).
commonly, can show only a photoconvulsive response on the EEG (355).
The mechanism for reflex seizures is unclear. The most attractive theory is that the attacks are caused by hyperexcitability of the sensory cortex, perhaps combined with a lack of cortical inhibition for a certain type of afferent input.
Treatment consists of avoiding the specific stimulus that induces seizures and administering anticonvulsants, preferably valproate or clonazepam. It is of note that the frequency of the TV or video game screen is important in provoking a seizure, with a 100-Hz screen being significantly safer than a 50-Hz screen. In addition, the distance of the child from the screen also is important, in that 1 m is safer than 50 cm (354).
Diagnosis
The diagnostic process in a child with epileptic seizures has two phases: the ascertainment of the type of seizure and its focus, if any, and an attempt to understand the cause for the attacks. Evaluation of the child with the first nonfebrile seizure has been reviewed by Hirtz and coworkers (356). They recommend the following steps: (a) determine whether a seizure has occurred, and (b) determine the cause of the seizure.
A thorough history, taken not only from the parent but also from the child, is crucial for arriving at a diagnosis. Usually, the physician is not able to witness an attack, and hospitalizing the child in the hopes of recording a seizure is financially prohibitive, although in some instances, it ultimately may be necessary.
Differential Diagnosis
The diagnosis of a generalized tonic-clonic seizure is usually made without much difficulty, although psychogenic seizures (hysteric seizures) also should be kept in mind despite their current relative rarity (357). The best discussion of the differential diagnosis between the two conditions is by Gowers, who emphasized the following points (48):
In the hysteric convulsion, the aura is absent or consists of palpitation, malaise, or a choking sensation.
During a hysteric attack, consciousness is impaired rather than lost.
The movements accompanying a hysteric seizure are somewhat coordinated but lack the definite sequence of a true tonic-clonic seizure. Tonic spasms are long and severe, often associated with opisthotonus or brief and irregular clonic movements.
Micturition and tongue biting are rarely seen in a hysteric attack.
The hysteric attack terminates suddenly, and the patient often resumes his or her former activities.
To these criteria, distinguishing hysteric seizures, proposed more than 100 years ago, can now be added the absence of abnormalities on EEG or video-telemetry EEG recordings during a hysteric attack (357,358).
Childhood hysteria can be encountered as early as 6 years of age. Although it is still more common in girls, in children younger than 10 years of age, the sexes are affected equally (359).
Lazare has listed several psychological criteria for the diagnosis of a conversion symptom, such as a hysterical seizure (360):
A disorder of somatization (i.e., long-standing psychosomatic symptoms affecting several organ systems)
An associated psychopathology (e.g., depression, personality disorder, schizophrenia)
A model for the symptom, based on the patient’s own previous illness or on that of an important figure in the patient’s life
Less important criteria for the diagnosis of a conversion reaction are emotional stress before the onset of symptoms, a disturbed sexuality (i.e., a history of seduction or incest), and the patient’s being the youngest child. These considerations do not always allow a definite diagnosis, and it is important to remember that casual evaluation can lead to an erroneous diagnosis.
Grand mal attacks also should be differentiated from syncope. Syncope is rare in childhood, being more common in adolescence, particularly in girls. The attacks usually occur in the upright position and are preceded by fatigue or emotional stress. Syncopal attacks are occasionally terminated by a brief generalized tonic or tonic-clonic convulsion, probably the result of cerebral anoxia. An EEG recording during a syncopal attack, if available, shows diffuse electric slowing rather than seizure activity (361).
Another condition requiring differentiation from grand mal attacks is breath-holding. Breath-holding spells are limited to children younger than age 6 years. They are precipitated by crying in response to emotional upsets. The attack is usually brief and accompanied by intense cyanosis or pallor. The child can be limp or opisthotonic. A short clonic convulsion induced by cerebral anoxia can terminate the spell.
Tharp has listed a variety of other disorders that mimic seizures (Table 14.9) (362).
A few comments with respect to some of the more commonly encountered diagnostic problems follow.
The differentiation between a prolonged absence seizure with motor accompaniments and a brief complex partial seizure should take the following items into account: that a complex partial seizure lasts longer and includes a greater variety of movements; that absence attacks do not have an aura, occur far more frequently than psychomotor seizure, and can often be elicited by
hyperventilation; that the patient with a complex partial seizure has partial clouding of consciousness for a brief period after the seizure has ended; and that the interictal EEG abnormalities in absence epilepsy are generally diagnostic.
hyperventilation; that the patient with a complex partial seizure has partial clouding of consciousness for a brief period after the seizure has ended; and that the interictal EEG abnormalities in absence epilepsy are generally diagnostic.
TABLE 14.9 Episodic Disorders that Mimic Seizures | ||
|---|---|---|
|
Pavor nocturnus (night terror) should be distinguished from nocturnal complex partial seizures or major motor seizures. Pavor nocturnus is a paroxysmal sleep disturbance occurring during arousal from slow-wave sleep. The child appears agitated or leaves bed crying and in apparent terror. Heart rate and respiratory rate are increased. The episode is brief, often lasting less than a minute, and the child returns to sleep. Generally, efforts to rouse the child fail, and the child has no recollection of the attack. The EEG during pavor nocturnus is normal (363), and children generally outgrow this condition (see Chapter 15).
Myoclonic seizures of the body or extremities commonly occur when a child drops off to sleep and in a few families can be unusually violent. The syndrome of severe nocturnal myoclonus, although benign and not amenable to drug therapy, must be differentiated from a true nocturnal epileptic seizure (364).
Tics are usually distinguished from myoclonic seizures because the patient is able to control tics voluntarily, they consist of the same movement each time, and they occur in patients who manifest other evidence of psychological disturbance and are aggravated by emotional strain.
Spasmus nutans (see Chapter 8) is often confused with epileptic seizures, particularly salaam seizures. Unlike in salaam seizures, children with spasmus nutans do not lose consciousness during an attack, and intellectual development remains normal. Vertical or horizontal nystagmus affecting one or both eyes is a common feature of spasmus nutans. Head thrusts and blinking observed in congenital ocular motor apraxia have been mistaken for the seizure manifestations of Lennox-Gastaut syndrome (365).
Patient History
Because the diagnosis of a seizure disorder relies to a great extent on the history furnished to the physician, an intentionally falsified history leads to the erroneous diagnosis of epilepsy. Over the last decades, we have seen an increasing number of children with Munchausen disease by proxy who have been brought in with a history of a seizure disorder or recurrent apnea (366). Fictitious epilepsy, the term used by Meadow, is a not at all rare form of child abuse and can lead to years of unwarranted invalidism (367). Its diagnosis is difficult but should be considered when, in addition to epilepsy, the youngster suffers from other disorders equally difficult to document. These include diarrhea and food allergies, hematuria and hematemesis, and apneic episodes. Additionally, the patient often has an inexplicable mixture of overdosing and undertreatment, as verified by blood anticonvulsant levels. When fictitious epilepsy is suspected, verification of the history from individuals outside the family (e.g., teachers) and observation in the hospital are indicated.
Physical Examination
The physical examination in a patient with a seizure disorder can be abnormal when the patient has underlying cerebral pathology (Fig. 14.7). A small fraction of children with long-standing seizure disorders unaccompanied by neurologic abnormalities harbor a cerebral tumor. Although
imaging studies, especially MRI, disclose the neoplasm, it is impractical and far too costly to subject every child who has experienced a seizure to imaging studies. What makes the diagnosis even more difficult is the fact that in most of the children with an underlying tumor, neither the type of seizure nor the initial EEG suggests a focal disorder; even a CT scan performed as part of an initial evaluation can be normal. Page and associates compiled the clinical features that they consider indicative of a cerebral tumor in children with a seizure disorder (Table 14.10) (368). The physician must keep in mind that the physical examination also can be abnormal when the patient is examined shortly after a seizure, and when cerebral dysfunction results from recurrent seizures or is the consequence of anticonvulsant medication.
imaging studies, especially MRI, disclose the neoplasm, it is impractical and far too costly to subject every child who has experienced a seizure to imaging studies. What makes the diagnosis even more difficult is the fact that in most of the children with an underlying tumor, neither the type of seizure nor the initial EEG suggests a focal disorder; even a CT scan performed as part of an initial evaluation can be normal. Page and associates compiled the clinical features that they consider indicative of a cerebral tumor in children with a seizure disorder (Table 14.10) (368). The physician must keep in mind that the physical examination also can be abnormal when the patient is examined shortly after a seizure, and when cerebral dysfunction results from recurrent seizures or is the consequence of anticonvulsant medication.
 FIGURE 14.7. Visual field defect in child with complex partial seizures and temporal lobe tumor. The child has a superior quadrantal hemianopia with sparing of the macula. (OD, right eye; OS, left eye.) (Courtesy of Dr. R. Hepler, Department of Ophthalmology, University of California, Los Angeles, UCLA School of Medicine.) |
TABLE 14.10 Warning Signs of Cerebral Neoplasms in 23 Children with Long-Standing Seizure Disorder and Brain Tumor | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
TABLE 14.11 Neurologic Complications of Commonly Used Antiepileptic Drugs | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The patient may be confused following a severe seizure, and transient neurologic signs, including intention tremor, incoordination, weakness of one or more extremities, and pathologic exaggeration of reflexes, are common. When convulsions recur at brief intervals, these signs can persist for prolonged periods.
Cerebral damage can result from recurrent seizures. Usually, this damage takes the form of intellectual deterioration and emotional disturbance. The incidence of these complications is treated in the section on Prognosis.
A number of anticonvulsants can induce neurologic abnormalities (Table 14.11).
Laboratory Tests
Laboratory studies are directed toward uncovering the cause of the seizures. Various diagnostic procedures are customarily performed on patients with seizures who lack a history or neurologic findings that point to a diagnosis.
Blood Chemistries
Serum glucose and calcium levels are obtained for all infants with seizures and for older children whose histories raise the possibility of a metabolic disturbance (see Chapter 17). In neonates, an evaluation of renal function also is indicated. Serum electrolyte disturbances are rare in patients with recurrent seizures, but hyponatremia (sodium of less than 125 meq/l) was associated with seizures in 70% of infants under 6 months of age (356). In children with seizures owing to hypernatremia, the history
usually suggests a fluid imbalance. A toxicology study also is indicated.
usually suggests a fluid imbalance. A toxicology study also is indicated.
Electroencephalography
The EEG can be useful in determining the type of seizure the patient experiences. It differentiates between absence and complex partial seizures, and between absence attacks and atypical petit mal. Occasionally, it can assist in establishing a diagnosis when the history is inadequate or not diagnostic. It also is the best predictor of the likelihood of recurrence. The EEG must be interpreted in conjunction with the patient’s history. A normal EEG, seen in approximately one-half of epileptic patients, does not exclude the diagnosis of epilepsy, nor does an abnormal EEG necessarily establish it. King and colleagues have found that it is easier to differentiate between a primary generalized and a secondarily generalized seizure disorder if the initial EEG is performed within 24 hours of the seizure. They also recommend that should the initial EEG be normal, a second, sleep-deprived EEG should be performed (369). Hirtz and colleagues also state that an EEG obtained within 24 hours of a seizure is more likely to show epileptiform abnormalities and can guide the physician as to whether neuroimaging is necessary (356).
Electric abnormalities can be seen during an overt attack as well as between seizures. Generalized tonic-clonic seizures arising from subcortical excitation can be recorded on the EEG, but the abnormalities are usually obscured by movement artifacts. One exception is the major seizure with a focal cortical origin. In this condition, random focal spiking or spike-wave discharges that had already been seen during the interseizure period increase in frequency and amplitude and spread both contiguously and via subcortical centers. Generalization can occur within a fraction of a second after the onset of the focal seizure; less commonly, it develops slowly over as long as 20 seconds. Following the seizure, electric activity is depressed. Recovery starts with slow waves, normal cortical activity first appearing contralateral to the epileptic focus, then ipsilateral, and finally over the focus itself.
Less commonly, the ictal tracing is marked by suppression of electrical activity, or low-voltage fast activity, an indication of disinhibition.
The EEG during an absence attack shows bilateral high-voltage synchronous alternating spike-wave complexes, most commonly 3 to 4 Hz (see Fig. 14.3). The discharge frequency can be increased by hyperventilation. As the discharge continues, the frequency of the spike-wave complexes tends to slow down.
In most patients with complex partial seizures, the EEG patterns accompanying the seizure are slow, rhythmic, and usually bilaterally synchronous, 4- to 6-Hz discharges, most prominent in the frontal and temporal areas but becoming generalized as the seizure progresses. At the conclusion of the attack, the EEG is featureless, and recovery of normal activity is delayed for many minutes.
The interseizure record in patients with major motor attacks can be normal or can demonstrate a number of nonspecific abnormalities, including disorganization, spike discharges, loss of a-rhythm or loss of the highest expected frequency activity for the child’s age, and, occasionally, continuous or intermittent focal slowing. This last finding, particularly when it is continuous, should always raise the possibility of an underlying tumor or other structural lesion and indicates the need for imaging studies.
In complex partial seizures, the interseizure tracing is normal or shows nonspecific abnormalities or spike foci, which usually arise from the anterior portions of the temporal lobes (Fig. 14.8). Often, the seizure focus is apparent only during the transition between wakefulness and sleep, and a combined wake and sleep tracing should always be obtained from children suspected of having complex partial seizures. In approximately 80% of children with this condition, three serial wake and sleep recordings uncover an abnormality. In a number of patients, secondary spike foci can be recorded from the opposite temporal lobe. It is rare to find focal temporal spike discharges in children younger than 8 years of age, even in those with a classic clinical picture of complex partial seizures.
 FIGURE 14.8. Right anterior temporal spike discharge in patient with complex partial seizures. (C, central; F, frontal; FP, frontopolar; O, occipital; P, parietal; T, temporal.) (Courtesy of Dr. Gregory Walsh, Department of Neurology, University of California, Los Angeles, UCLA School of Medicine.) |
Atypical spike-wave discharges (i.e., spike-wave discharges at a frequency less than 2.5 Hz) and polyspike-wave discharges are common in children with Lennox-Gastaut syndrome. They indicate a poor prognosi in terms of seizure control and normal intellectual function. The high positive correlation between hypsarrhythmia and infantile spasms has already been discussed.
With EEG maturation, the EEG in patients with seizure disorders can undergo significant changes. Focal spike activity tends to migrate from the occipital to the midtemporal and ultimately to the anterior temporal leads (370). In patients with 3-Hz generalized spike-wave discharges, an improvement in clinical status can be associated with the disappearance of the abnormal EEG pattern, which is often replaced by a wandering spike focus (371). In patients with focal seizure discharges, mirror spike activity can appear. In some, this second focus is suppressed by anticonvulsants; in others, it becomes the only active focus (371).
For further information on the clinical aspects of EEG, the reader is referred to one of the several standard EEG texts.
Lumbar Puncture
Lumbar puncture is not performed routinely in patients with seizure disorders. Because a significant proportion of infants with bacterial or tuberculous meningitis have a febrile seizure as their initial symptom (see Chapter 7), we believe a lumbar puncture is indicated in patients who have experienced their first febrile seizure and in all infants having their first seizure.
Although the CSF is by definition normal in the majority of patients with idiopathic epilepsy, minor abnormalities were found in the classic study of Lennox and Merritt (372). In 4% of their patients, pleocytosis was slight (5 to 10 cells/μL), and in 10%, protein content was increased (45 to 85 mg/dL). Both abnormalities were believed to be related to the presence of small areas of cerebral contusion that resulted from a fall attending the seizure. They had disappeared when the spinal tap was repeated several weeks later. More recent studies are consistent with these observations (373,374). Wong and coworkers, who examined the CSF of children within 24 hours of a seizure, found a normal range of 0 to 12 cells in infants under 4 months of age, and a range of 0 to 8 cells in infants over 4 months. The normal postictal protein was less than 100 mg/dl in infants less than 2 months, less than 60 mg/dl between 2 and 4 months, and less than 35 mg/dl in infants over 4 months of age (375). We suspect that abnormalities in cell count and protein level, when present, reflect the cerebral edema, disturbed autoregulation, and the breakdown of the blood–brain barrier that attend a prolonged seizure and that have been documented by MRI (376). In contrast to adults, seizure-induced neuronal injury is not marked by an increase in neuron-specific enolase; rather, increases reflect the presence of metabolic or genetic abnormalities (377).
Imaging Studies
MRI and CT scans are the procedures currently used in the initial evaluation of a child with a seizure disorder. The question as to whether an emergent imaging study should be performed as part of the evaluation of the child that presents with an afebrile seizure has been considered by several authors. In the experience of Maytal and coworkers, only 6% of children who presented with cryptogenic seizures had an abnormal CT. By contrast, the CT was abnormal in 60% of children whose seizures were considered to be symptomatic (378). Similar results were obtained by Sharma and coworkers (379). From studies such as these, it is evident that neuroimaging of children with the first afebrile seizure should be reserved for those who have conditions that place him or her at an increased risk for neuroimaging abnormalities. These include an abnormal neurologic examination, a history of malignancy, sickle cell disease, bleeding disorder, a closed head injury, or travel to an area endemic for cysticercosis (379). Infants under 33 months with focal seizures should undergo imaging as well. In this group, in the experience of Sharma and coworkers, CT abnormalities were seen in 29% (379). By contrast, only 2% of CTs were abnormal in children with nonfocal seizures and no predisposing conditions (379). Hirtz and colleagues found that the yield of abnormalities on CT scan is low in the presence of a normal EEG and a normal neurological examination, and that abnormalities when present do not influence treatment (356).
An imaging study is indicated for the child with recurrent seizures under several circumstances: in the presence of an abnormal neurologic examination, including dysmorphic features, or skin lesions suggestive of a phakomatosis (64%); in the presence of focal EEG abnormalities, particularly focal slowing (63%); with a history of neonatal seizures (100%); and with a history compatible with simple partial or complex partial seizures (52% and 30%, respectively). The numbers in parentheses represent percentage of positive CT scan results seen by Yang and coworkers in their series of seizure patients (380); the percentages of positive combined CT and MRI studies would undoubtedly have been higher.
Whether imaging studies also are indicated for children who by history and examination do not fulfill these criteria is a matter of individual judgment. Children whose neurologic examination and EEG are normal have a low yield of positive scan results (356), as do children with primary generalized seizures [5% and 8%, respectively, in the series of Yang and coworkers (380)]. In confirmation of this experience, King and coworkers found no MRI abnormalities in any of their patients with EEG-confirmed primary generalized seizures (369).
Several studies have compared CT scans and MRI for their respective advantages and disadvantages in evaluating a seizure patient. Although the two procedures occasionally provide complementary information, MRI is the preferred initial screening procedure. Unlike CT scanning, MRI detects congenital malformations of the cortex, gray matter heterotopias, and a large proportion of arteriovenous malformations, some of which can be angiographically occult. MRI also is an excellent screening test for detecting neoplasms, particularly when it is used with gadolinium enhancement. On the other hand, CT scans are more sensitive for small foci of calcification.
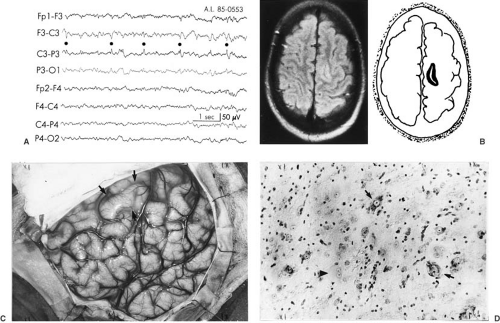 FIGURE 14.9. An 18-year-old girl with onset at 6 years of age of focal myoclonus involving right arm and shoulder and right hemiparesis. A: Representative section of electroencephalogram showing frequent sharp waves with phase reversals over the left central region (C3). (C, central; F, frontal; O, occipital; P, parietal.) B: Magnetic resonance imaging showing left hemiatrophy and an area of slightly increased signal corresponding to the abnormal gyrus (see adjacent diagram). C: Left cerebral hemisphere at operation. Note enlarged gyrus (black arrows). D: Microscopic section at junction of cortex and white matter showing disruption of cortical lamination with abnormally large dysplastic neurons (arrows) and large astrocytes (arrowhead) (Luxol fast blue, ×130). (Courtesy of Drs. R. Kuzniecky and F. Andermann, Department of Neurology, McGill University, Montreal, Canada.) |
The joint use of EEG and imaging studies has been applied most successfully in the evaluation of patients with focal seizure disorders. MRI is effective in detecting underlying abnormalities in the cortical architecture of children with simple partial epilepsies (369,381) (Fig. 14.9). In young children with intractable partial epilepsies, malformations of the cortex are detectable on MRI. These abnormalities are most common in the central cortical area, particularly in the region of the operculum. The complexity of cortical gyration makes difficult the detection of subtle abnormalities, and when MRI is correlated with pathologic examination of brain tissue resected from epileptic seizure foci, only approximately one-half of the malformations were detected before surgery by MRI (382). MRI results are abnormal in most patients with severe mesial temporal sclerosis as well as in a large proportion of subjects with pathologically proven mild to moderate sclerosis (383,384). These abnormalities are best demonstrated with T2-weighted or fluid-attenuated inversion recovery (FLAIR) images. Additionally, MRI can detect small epileptogenic lesions in the temporal lobe, notably hamartomas and low-grade gliomas (385,386). When children with newly diagnosed epilepsy were subjected to both MRI
and CT, both studies were abnormal in 83%, and in the remainder, the CT was normal but the MRI was abnormal. The MRI also found a number of incidental abnormalities, such as Chiari 1 malformations, pineal cysts, and arachnoid cysts (387).
and CT, both studies were abnormal in 83%, and in the remainder, the CT was normal but the MRI was abnormal. The MRI also found a number of incidental abnormalities, such as Chiari 1 malformations, pineal cysts, and arachnoid cysts (387).
 FIGURE 14.10. Positron emission tomography using labeled fluorodeoxyglucose. Interictal scan in a patient with complex partial seizures since age 5 years. There is a zone of relative hypometabolism in the left temporal lobe (t). The electroencephalogram on this patient showed a left mesial temporal spike focus. Computed tomographic scans showed possible enlargement of the left temporal horn, but angiographic and pneumoencephalographic studies were normal. Pathologic examination following left temporal lobectomy revealed a mesial temporal lobe sclerosis. (From Engel J, Crandall PH, Rausch R. The partial epilepsies. In: Rosenberg RN, et al., eds. The clinical neurosciences, Vol. 2. Neurology/neurosurgery. New York: Churchill Livingstone, 1983. With permission.) |
Transient abnormalities at the site of an actively discharging epileptic focus can be visualized by MRI, particularly on T2-weighted images (388). These abnormalities can reflect hyperemia or can be caused by a shift of water from the extracellular compartment into dendrites or glia (389). Similar changes also can be visualized on CT scans. The abnormalities take from 1 to 48 hours to develop and can resolve within 1 week or as late as 12 weeks (390). Additionally, some drugs, notably digoxin and heparin, can increase the signal intensity on MRI (391).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


