1 Pathology and Molecular Genetics of Common Brain Tumors
Introduction
This chapter aims to provide an outline of the surgical pathology and the recognized genetic and molecular changes of common tumors of the nervous system in children and adults. The current World Health Organization (WHO) histological classification for nervous system tumors will be used as its framework.1 The histological basis for classification and malignancy grading of the tumors is briefly presented and some of the common diagnostic problems outlined. The WHO classification is complex, listing over 120 histological entities. In the case of some of the tumors recognized, there is as yet only a histological description and no genetic information is available. The reader is referred to the fourth edition of the WHO classification of tumors of the nervous system and the specialized literature for the tumor types not addressed here.1 The genetic and molecular information we have on the common tumors is steadily increasing, but is still rudimentary. While the genetic and molecular findings are not, as yet used, clinically, as soon as molecular targeted therapies become available and are found to be effective, histological investigation will have to be supplemented with molecular data.
Most classifications of brain tumors presented during the last 60 years build on the 1926 work of Bailey and Cushing.2 In their classification, tumors were named after the recognized cell types in the developing embryo/fetus or adult that the tumor cells most resembled histologically. The cell type of origin of the majority of brain tumors is unknown, as no premalignant states are recognized. In some tumors the cells may be so dysplastic that they show no similarities to any normal cell type—thus the use of terms such as glioblastoma. In the present WHO classification (for an overview, see Table 1-1), tumors are divided up into those of neuroepithelial origin (includes the glial, glioneuronal, neuronal, pineal and embryonal tumors), tumors of cranial and paraspinal nerves, tumors of the meninges, lymphomas and hematopoetic neoplasms, germ cell tumors, tumors of the sellar region and metastases. The tumor types and their possible WHO grades are given in Table 1-1.

GENERAL CONSIDERATIONS
Many brain tumors are morphologically heterogeneous, and many brain tumor types are known to become more malignant with time, with their progression initially being focal. Thus, for both reasons, adequate sampling of a tumor is essential to determine the correct tumor classification or type of tumor as well as the WHO grade. Classification of brain tumors is dependent on the recognition of areas with the characteristic histology for a particular tumor type, often assisted by immunocytochemical methods. Immunocytochemistry permits the demonstration of antigens associated with a particular cell type and even their subcellular location. As yet, there are no single antibodies or even panels of antibodies that unequivocally identify any of the brain tumors listed in the 2007 WHO classification.2 Thus, the presence or absence of an antigen only adds a further piece of information, helping to indicate the tumor type.
Once a tumor has been classified, the histologically most malignant part of the tumor determines the malignancy grade (often referred to as the WHO grade when using the criteria defined by the WHO classification).1 The histological criteria for malignancy grading are not uniform for all tumor types and thus all tumors must be classified first before a WHO malignancy grade can be determined. The WHO system recognizes four grades of malignancy; these provide an assessment of the biological aggressiveness of the untreated tumor. Grade I tumors are the biologically least aggressive and may be cured by surgery alone (e.g., pilocytic astrocytoma). Grade IV tumors are biologically highly aggressive, with rapid growth and the ability to infiltrate locally and disseminate within the central nervous system. Untreated, they are rapidly fatal (e.g., glioblastoma). General criteria for determining WHO malignancy grade include cellularity, degree of polymorphism and atypia, the incidence of mitoses, the presence of spontaneous necrosis and the degree of angiogenesis induced by the tumor (microvascular proliferation), but, as indicated above, these are not universal. The criteria for determining the WHO grade for each tumor type have been empirically derived by correlating the histology of surgically removed tumor tissue with otherwise untreated patient survival. Extrapolation from such studies provides a basis for an assessment of prognosis, but the provision of therapy may radically alter this assessment.
The use of objective methods of measuring cell proliferation and cell death (apoptosis) in tumors to determine WHO malignancy grade is conceptually attractive. However, the wide variations in proliferation indices observed in different areas of individual tumors have resulted in difficulties in defining relevant proliferation levels. The same applies to the assessment of the numbers of cells undergoing apoptosis. In the WHO system, mitotic counts (mitoses per ten 0.16 mm2 high power fields) are currently only used in the grading of meningiomas. The MIB-1 antibody that recognizes the same antigen as the Ki67 antibody and thus cells in the cell cycle can also be used to assess cell cycle activity. Other antibodies that identify antigens associated with proliferation (e.g., Cdc6 and Mcm5) can be applied to formalin-fixed, paraffin-embedded tissues following microwave antigen retrieval.3,4 However, the WHO system generally only gives information on commonly observed ranges for both the mitotic index and MIB index for most tumor types and WHO malignancy grades.
Most brain tumors are sporadic. However, a number of familial cancer syndromes are associated with an increased risk of brain tumors (see Table 1-1 and the references therein). Even in the case of the commonest syndromes (neurofibromatosis type 1 and neurofibromatosis type 2), the precise relative risk is difficult to define.
In contrast to many epithelial neoplasms, no lesion is recognized as a precursor for any brain tumor type and, as a result, the cell of origin of these monoclonal proliferations is unknown in all cases. Recent work in animal models provides some data supporting the idea that some brain tumors arise from neuroectodermal stem cells. They are present throughout life, have proliferative potential, are migratory and can differentiate along a number of paths—all features they have in common with brain tumor cells. Furthermore, there is some evidence that at least some of the common types of brain tumors may be made up of a smallish population of therapy-resistant tumor-initiating cells (also called tumor stem cells in some texts), with the main bulk of the tumor consisting of a progeny lacking these tumor-initiating abilities.5–9 If this tumor-initiating cell population is not eradicated, the tumor will recur−a common experience with current therapies for many of the malignant brain tumors. Thus, much effort is currently being channeled into defining and studying this subpopulation in the hope of finding ways to specifically kill these cells.10,11 While the genetic status (e.g., gene copy number, mutations, amplifications, etc.) of tumor-initiating cells and their progeny is likely to be identical, the epigenetic status (e.g., methylation) and the expression characteristics may differ considerably.
Childhood Tumors
PILOCYTIC ASTROCYTOMAS
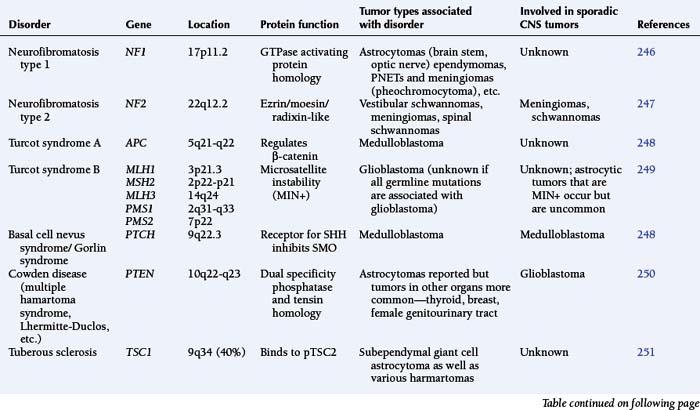
The astrocytomas encompass a number of tumors of differing grade including the pilocytic astrocytomas. The majority of astrocytomas are found in adults (see following discussion). The commonest form of astrocytoma found in children is the pilocytic astrocytoma, WHO malignancy grade I. These tumors may arise anywhere from the optic nerve to the medulla oblongata. They most commonly occur in the cerebellum. They may be solid, mono- or polycystic and are generally well circumscribed. The prognosis for patients with pilocytic astrocytomas that can be excised is relatively good, with over 90% 10-year survival reported.12 Pilocytic astrocytomas are generally biologically unaggressive, and, in contrast to the adult diffuse astrocytic tumors, maintain their grade I status over years and even decades. Only very occasionally will these tumors progress to malignant astrocytic tumors or seed the neuroaxis.13 Pilocytic astrocytomas show a wide morphological spectrum, from pilocytic bipolar cellular areas with Rosenthal fibers (Fig. 1-1A) to less cellular protoplasmic astrocytoma-like areas with eosinophilic granular bodies and clear cells. Such clear-cell areas are reminiscent of oligodendrogliomas and, in the posterior fossa, may also be confused with clear cell ependymomas, particularly in biopsies. The presence of features typically associated with a malignant biological behavior (e.g., atypia, microvascular proliferation (Fig. 1-1B), or even mitosis) does not carry the same sinister implications as in the adult diffuse astrocytic tumors. This morphological variability can make histopathological diagnosis extremely difficult. NF1 patients have an increased incidence of pilocytic astrocytomas, particularly involving the optic nerve, and these behave in a particularly benign fashion.14
Many cases of pilocytic astrocytomas have been studied cytogenetically and further cases analyzed using conventional comparative genomic hybridization (cCGH). Many show normal cytogenetic and cCGH findings.15–18 Polysomy has been found, most commonly of chromosomes 5 and 7, and, in addition, of chromosomes 6, 11, 15, and 20 (in decreasing frequency). Polysomy has been reported to be most common in adult patients with pilocytic astrocytoma.19 Molecular genetic studies have been few; single TP53 mutations have been reported, and loss of one allele of NF1 has been found in pilocytic tumors from NF1 patients but not in sporadic cases.20–24 Studies of promotor methylation of some genes known to be involved in adult diffuse astrocytic gliomas have provided inconsistent data.25,26 There appears to be no evidence for methylation of the NF1 gene in sporadic tumors.27 Recently a number of groups have noted that a small region on 7q34 has increased copy number in a high percentage of pilocytic astrocytomas.28–30 Further studies by Jones et al. showed this to be a tandem duplication of approximately 2 Mb resulting in an in-frame fusion gene encoding the kinase domain of the BRAF proto-oncogene. The fusion protein was shown to have constitutive kinase activity and to transform NIH3T3 cells. This rearrangement produced a specific and unique fusion BRAF oncogene in 29/44 (66%) of the pilocytic astrocytomas. An additional two tumors showed the well-known BRAF V600E mutation and a further three tumors were in patients diagnosed with the NF1 syndrome. Thus, a total of 34/44 (77%) of the pilocytic tumors in the series showed alterations in the MAPK (mitogen-activated protein kinase) pathway.29 This tandem duplication producing a fusion oncogene was not found in over 200 high-grade astrocytic tumors, demonstrating the specificity of the finding. As this rearrangement occurs in the majority of pilocytic astrocytomas, it provides a diagnostic marker for this tumor type as well as a potential target for molecular therapies.
EPENDYMOMA
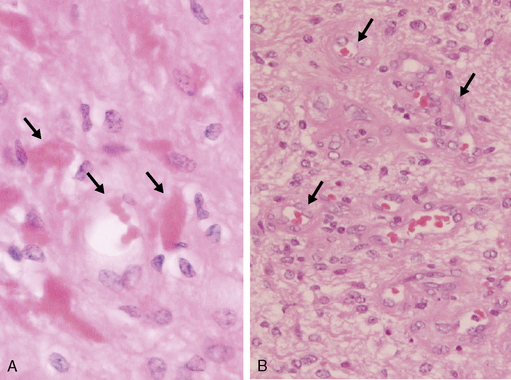
Ependymomas arise at or close to ependymal surfaces (ventricular system and the spinal canal) and very occasionally at extraneural sites. The commonest location is in the fourth ventricle, followed by the spinal canal (in adults), the lateral ventricles, and the third ventricle. The formation of ependymal rosettes and sometimes canals (Fig. 1-2), in some area of the tumor is a key histological feature. More commonly, perivascular pseudorosettes are found; however, these are not specific to ependymomas and can occur in other gliomas. As indicated above, the differentiation of ependymomas (WHO grade II) from anaplastic ependymomas (WHO grade III) is not well defined and is usually based on low mitotic rate, low levels of cellularity and nuclear polymorphism. Necrosis and microvascular proliferation do not have the same significance in this tumor type as in the adult astrocytic tumors described later in this chapter. Most ependymomas (WHO grade II) show immunoreactivity for glial fibrillary acidic protein (GFAP), S-100 protein, and epithelial membrane antigen (EMA). There have been a number of immunocytochemical studies attempting to identify prognostic markers. Some have been shown to be significant in single reports, but require confirmation.31–33 Among these, the expression of human telomere reverse transcriptase (hTERT) has been recently reported as a predictive marker of poor outcome in pediatric intracranial ependymomas.34 The cell of origin of these tumors has been suggested to be the radial glia cell.35
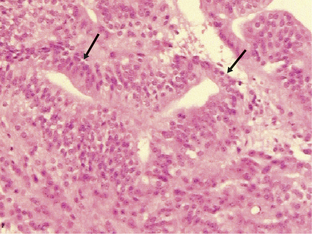
Figure 1-2 Ependymoma WHO grade II. Note the multiple ependymal canals (arrows) lined by tumor cells resembling a normal ependymal surface. Note that the tumor is relatively cellular (compare with the diffuse astrocytoma WHO grade II, Fig. 1-4A). (H&E)
Classical cytogenetic, metaphase, and array CGH studies as well as molec-ular genetic studies have identified copy number abnormalities that affect almost all chromosomes in the majority of ependymoma types.20,36–47 Even the often incidentally discovered and very benign subependymomas show copy number changes involving, in some cases, chromosomes 6, 7, 8, and 14 in some cases.46 Myxopapillary ependymomas frequently show concurrent gain of chromosomes 9 and 18.41 Deletions and regions of increased copy number are common in the ependymomas WHO grade II and the anaplastic ependymomas WHO grade III. Losses on chromosome 22 were an early finding (20) and this has been shown to be a frequent event, particularly in adult spinal ependymomas (over 50%). This is true both of ependymomas arising in patients with the neurofibromatosis Type 2 (NF2) syndrome as well as in sporadic cases, but is less frequent in pediatric and intracranial ependymomas.42,48 Loss of both wild-type copies of the NF2 gene has been demonstrated in both the sporadic and NF2 syndrome-related intramedullary spinal ependymomas of adults.37,49,50 This occurs in the NF2 patients by loss of the single wild-type NF2 gene with retention of the constitutively mutated copy, while in the sporadic cases it is generally by loss of most of one 22 q and somatic mutation of the single retained NF2 gene.37,50–53 Some ependymomas have been reported to have no apparent abnormali- ties, but this probably reflects on the level of resolution obtained with the analytical method used. The genes targeted by the allelic losses and gains in ependymomas are in most cases unknown. Some studies have attempted to correlate chromosomal abnormalities with progression-free survival or overall survival. The findings to date indicate that gain of 1 q is associated with a worse clinical outcome.38,42,54 Gains of 7 q and 9 p as well as losses of 17 and 22 have been reported to occur more frequently in recurrent tumors.55 Single cases have been reported with loss of the wild-type allele of genes such as the MEN1 gene.50,56 Mutations of TP53 or genetic changes affecting the integrity of the p53 pathway are uncommon in ependymomas. This contrasts with their frequency in the diffuse astrocytic tumors.57–60 More recently, global gene expression has been analyzed in ependymomas; preliminary findings suggest that there are specific expression profiles associated with the various histological subtypes and even patient survival.43,61,62 Most of these studies have been done on relatively small series of tumors and much further work is required before any of the findings can be introduced into clinical practice.
MEDULLOBLASTOMA
Medulloblastomas are highly malignant tumors and are WHO graded as IV. They have a peak incidence in childhood (around 7 years of age) but can occur into late middle age. Childhood and adult medulloblastomas are histologically identical being highly cellular, malignant, invasive tumors occurring in the posterior fossa. There are a number of subtypes; these include the classic, desmoplastic/nodular, large cell, and anaplastic cell variants. The large cell and anaplastic cell variants can be difficult to differentiate, but both have a significantly poorer prognosis than the other subtypes and therefore need to be recognized. All subtypes can show production of melanin and focal myogenic differentiation. Medulloblastomas in children (particularly the large cell variant) must be differentiated histologically from atypical teratoid or rhabdoid tumors, which have an extremely poor clinical outcome and do not respond to the current relatively successful treatment protocols for medulloblastomas.63–67 Loss of wild-type INI1/hSHF5/SMARCB1 genes is the genetic hallmark of the atypical teratoid or rhabdoid tumors.68,69 Classical medulloblastomas consist of densely packed tumor cells with round to oval or carrot-shaped hyperchromatic nuclei with scanty cytoplasm, high mitotic and apoptotic rates, and usually neuroblastic rosettes in some areas (Fig. 1-3). In adults, the possibility of a metastasis of a small-cell lung cancer must often be excluded. Neuronal differentiation and glial differentiation may be present, particularly in the nodular areas of the desmoplastic/nodular and medulloblastoma with extensive nodularity variants. Microvascular proliferation is uncommon. Tumors arise with similar frequency in the cerebellar vermis (mainly in children) and the cerebellar hemispheres (older patients) and often invade the fourth ventricle, with occasional brainstem involvement. There is a high risk of seeding through the subarachnoid space due to the tendency of the tumor to penetrate the ependymal surface. Many antigens can be identified focally in medulloblastomas (nestin, vimentin, neurofilament proteins, GFAP, retinal S-antigen, N-CAMs, Trk-A, -B, -C, etc.) and while most are not of any great importance in the diagnosis of classical cases, the identification of EMA and smooth muscle actin will help differentiate atypical teratoid tumors from the large cell medulloblastomas.
Immunocytochemically-identifiable prognostic markers in medulloblastoma have been reported. High nuclear expression of p53 and high expression levels of Erb-B2 (Her2) have been reported as indicators of a poor outcome.70–77 Studies of TrkB mRNA levels have indicated that high levels are associated with a favorable outcome, however, some immunocytochemical studies of TrkB have not shown any differences between patients with tumors expressing the protein and those without.78,79 There have also been a number of gene-expression profiling studies of medulloblastomas and these, by correlating the data with outcome, have attempted to identify expression signatures indicating a good or bad prognosis.80,81
At the genetic level, gain of 17 q and loss of 17 p are common findings. Generally, this is associated with the formation of isodicentric 17 q (previously inaccurately called isochromosome 17 q). Isodicentric 17 q consists of two 17 q with two centromeres and two small fragments of proximal 17 p fused at the terminal ends of the 17 p sequences.82,83 The fusion occurs in a number ways in a region of complex repeat sequences.82 Isodicentric 17 q is observed in 30% to 50% of cases. Other frequent copy number aberrations reported are gain of the whole of 1 q, gain of chromosome 7, and loss of 10 q (84–89). Amplification of each of the three MYC genes has been found in individual cases; this is associated with a poor survival.82,90,91 Other genes found to be amplified in individual cases include PDGFRA, KIT, OTX2 (14q21-q22), and MYB.82,92,93 Various candidate genes have been examined in the common regions of loss including TP53, HIC1, and KCTD11 on 17 p and DMBT1, PTEN, and SUFU on 10 q. Only single cases with TP53 mutations have been found but a significant incidence of methylation of the HIC1 gene has been reported. DMBT1 has been identified as a deletion polymorphism in humans and the losses are therefore unlikely to be of significance. Single mutations of PTEN have been documented, and while constitutive SUFU mutations have been found in some children with medulloblastoma and seem to predispose to medulloblastoma in mice, no mutations have been identified in sporadic medulloblastomas.94–98 Metastatic disease has been reported to be associated with elevated expression levels of PDGF receptors and ligands by tumor cells.99,100
The study of two familial tumor syndromes exhibiting a predisposition to medulloblastoma formation has led to major advances in our understanding of medulloblastoma biology. Gorlin syndrome (also known as hereditary nevoid basal cell carcinoma syndrome) and Turcot syndrome type A (associated with the familial adenomatous polyposis [FAP] syndrome) are due to inherited mutations of one copy of the PTCH (9 q) and the APC (5 q) genes respectively. The protein products of these two genes are involved in two interconnected cellular pathways that are fundamental to neural development and cell turnover. Hemizygous loss and mutation of the retained allele of PTCH in sporadic medulloblastomas has been demonstrated.101,102 Mutations in other genes coding for components of the PTCH pathway have also been reported, if only in single cases. These include SMO103–105 and SUFU.95,98 All these mutations result in activation of the pathway, with increased transcription of a group of genes including WNT (see following discussion). Thus, inhibitors of this pathway could be therapeutically useful. Cyclopamine is a natural alkaloid that inhibits the PTCH pathway; this and other inhibitors are currently being investigated.106,107 A key protein in the Wnt pathway is APC, and it is the inheritance of a mutated APC gene that causes FAP syndrome. In sporadic tumors, only a few APC mutations have been identified. APC forms a complex with at least seven proteins that bind hyperphosphorylated β-catenin, permitting its ubiquitination and thus targeting β-catenin for degradation. This mechanism is important for the control of cellular levels of β-catenin. Tumor-specific mutations affecting the region of β-catenin that is phosphorylated (a requirement for sequestration by the APC complex) and the ubiquitin-binding region have been reported.108,109 This would allow cellular β-catenin to escape this control mechanism and to accumulate and act as a transcription factor. Mutations of AXIN1, a gene coding for one of the proteins in the APC complex, have also been found.110 Thus, disruption of these complex pathways, upstream or downstream of WNT, is associated with the development of medulloblastoma. This also clearly demonstrates that a cellular mechanism can be disrupted by mutations of many different genes coding for the proteins involved, indicating that it is the pathway that is the target in oncogenesis and not a particular gene. Further examples of this will be seen later in this chapter. Despite these advances, alterations in the genes coding for components of the PTCH and WNT/APC pathways have only been found in a small percentage of sporadic tumors.111,112 However, the WNT signaling is extremely complex and many aspects have yet to be investigated.113
Common Adult Tumors
DIFFUSE ASTROCYTIC TUMORS
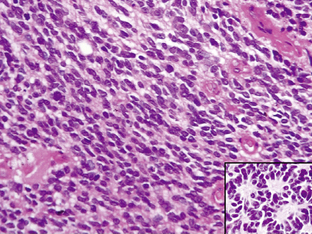
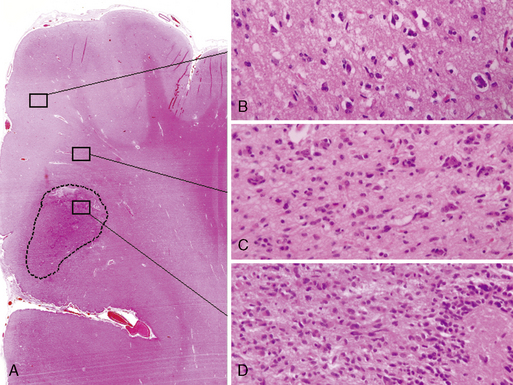
The diffuse astrocytic tumors include the astrocytomas (WHO grade II), the anaplastic astrocytomas (WHO grade III), as well as the glioblastomas (Fig. 1-4). These tumors predominate in adults, with the most malignant form, the glioblastoma WHO grade IV being the most common. The astrocytoma WHO grade II tumors (A) have a peak incidence in people between 25 and 50 years of age, while the peak incidence of glioblastomas (GB) is in those between 45 and 70. All are commoner in males and most are located in the cerebral hemispheres. The astrocytomas (WHO grade II) and anaplastic astrocytomas have been well documented to progress to tumors of higher malignancy grade.114,115 Glioblastomas are therefore divided into those that develop by progression from a previously diagnosed tumor of lower malignancy grade and those that appear to develop de novo.116,117 Both clinical and molecular data support the hypothesis that although these tumors may arise due to the mutation of different genes, the gene mutations target and disrupt the same cellular pathways.60,118–120 The relevance of the histolog-ically based malignancy-grading scheme is demonstrated by its prognostic value. Patients with a diffuse astrocytoma (WHO grade II) have an median survival of between 6 and 7 years, patients with anaplastic astrocytomas (WHO grade III) have a median survival half that time,121 while glioblastoma (WHO grade IV) patients have an average survival of a little over 1 year using the latest therapeutic regimens.122,123 The combination of radiotherapy with temozolomide, following debulking surgery, has improved outcome from a median survival in this group of 9 to 11 months, when treated with conventional surgery followed by radiotherapy and various chemotherapy regimens, to 14.6 months.123 As the term “diffuse astrocytoma” implies, these tumors do not have a clear border and show varying levels of infiltration into the surrounding brain (Fig. 1-5 A-D). Infiltrative ability varies widely from astrocytic tumors that are relatively well localized (rare) to gliomatosis cerebri (also rare), where there is extensive infiltration of a large region of the central nervous system.
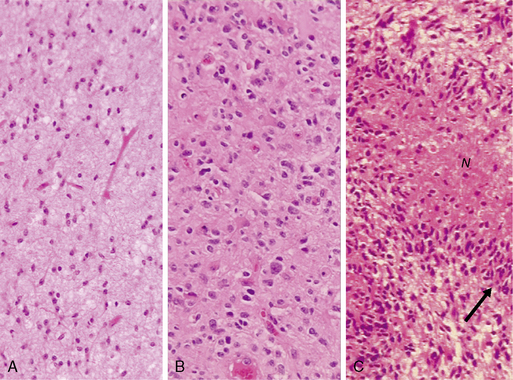
In diffuse astrocytomas (WHO grade II) the tumor cells morphologically resemble astrocytes, generally show little nuclear atypia, and have extensions producing a loosely textured matrix (Fig. 1-4A). They usually express S-100 protein and glial fibrillary acidic protein. Anaplastic astrocytomas (WHO grade III) are more cellular, with pleomorphic tumor cells showing nuclear atypia. There is some mitotic activity, but the tumor cells generally still display the histological and immunocytochemical characteristics of astrocytes (Fig. 1-4B). No evidence of spontaneous tumor necrosis or abnormal microvascular proliferation is permitted in anaplastic astrocytomas. Glioblastomas (WHO grade IV) are more cellular than the anaplastic astrocytomas and show a wide spectrum of morphologies. They can be very pleomorphic with giant-cell forms, but generally retain some of the phenotypical characteristics of astrocytes. Mitosis, spontaneous tumor necrosis with pseudopalisading of tumor cells, as well as florid endothelial proliferation are inevitably found in some areas of a well-sampled tumor (Fig. 1-4C). A large central necrotic area with a ring-like zone of contrast enhancement, representing the viable tumor tissue, can often be identified by neuroimaging.
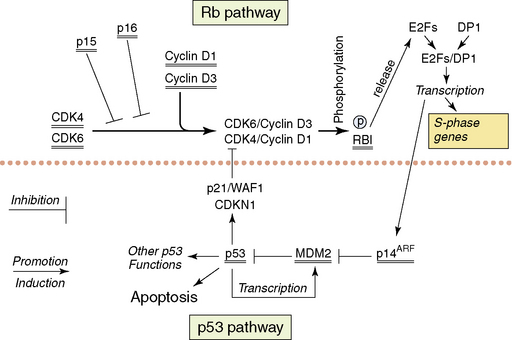
The following section will describe the major genetic abnormalities known in all three grades (II–IV) of diffuse astrocytomas. A simplified overview of the function of some of the proteins encoded by the genes referred to in the text is provided in Figs. 1-6 and 1-7. Cytogenetic and molecular data is limited on the diffuse astrocytomas (WHO grade II), as they are less common.15,124,125 Over 60% of these tumors have loss of alleles on 17 p including the TP53 locus, and the retained TP53 allele is mutated in the majority of cases.60,126,127 The absence of wild-type p53 is therefore the commonest abnormal finding,60,128 resulting in a nonfunctional p53 pathway. A small percentage of tumors have mutations of one allele but retain one wild-type allele. As the p53 protein is believed to function as a tetramer, and as tetramers with one abnormal p53 protein may not function normally, the finding of single mutated alleles together with a wild-type allele may be significant. Other genes coding for components of the p53 pathway (see Fig. 1-6), MDM2 and p14(ARF) have been studied in small numbers of tumors and no abnormalities have been reported. Studies of the TP53 related gene, P73, have not identified any mutations.129 Loss of alleles from 6 q, 13 q, and 22 q occur in some diffuse astrocytomas (WHO grade II). In tumors with LOH at 13q14.2 that encompass one copy of the RB1 gene, there is no evidence of mutation of the single retained gene copy.130 The same is true for the losses from 22 q encompassing one NF2 tumor suppressor gene.131,132 Deletion mapping of chromosome 6 shows losses on 6 q in a significant number of diffuse astrocytomas WHO grade II.133 The potential tumor suppressor genes in all of these regions remain unknown. There are no consistently reported amplified genes or amplified regions of the genome in these astrocytomas.130,134–137
High-level expression of the PDGF ligands and receptors has been observed in all grades of astrocytic tumor, suggesting the presence of autocrine/paracrine loops.138–141 Intracranial injection of a retrovirus containing PDGF-B induced glioblastomas in mice, suggesting involvement of the PDGF system in the pathogenesis of astrocytic tumors.142 Overexpression is not associated with amplification except in the case of the PDGFA receptor gene (PDGFAR), which has been reported to be amplified and overexpressed in small subset of glioblastomas.143–145
Epigenetic changes such as hypermethylation of tumor suppressor gene promoters have only recently received attention. There is some evidence that some tumor suppressors are methylated in diffuse astrocytomas WHO grade II (e.g., the PTEN gene).146 However, there are likely to be many other genes affected by aberrant methylation in the case of the astrocytomas WHO grade II.147,148 The changes found in the astrocytomas WHO grade II form the baseline for studies of the genetic abnormalities associated with progression in the adult diffuse astrocytic tumors.
The genetic data on the anaplastic astrocytomas (WHO grade III) is also limited. Loss of one copy and mutation of the retained copy of TP53 occur at approximately the same frequency as in the diffuse astrocytomas (WHO grade II).60 Thus, the p53 pathway is nonfunctional in the majority of cases (more than 60%). Cytogenetics, comparative genomic hybridization, and molecular genetic techniques all show that losses of genetic material from 6 q, 13 q, 17 p, and 22 q, as seen in the WHO grade II astrocytomas, occur at similar or higher frequencies in anaplastic astrocytomas. With the sole exception of copy number abnormalities of 19 q probably representing extra copies (targeted gene[s] unknown), there are no conclusively demonstrated abnormalities specific to this WHO grade. Around 20% of anaplastic astrocytomas show similar genetic abnormalities to those found in glioblastomas, involving other components of the p53 pathway (i.e., MDM2 and p14(ARF)) and leading to disruption of the Rb1 pathway (Fig. 1-6); these are discussed in the glioblastoma section.60
The majority of glioblastomas arise de novo; this has ensured their study in considerable numbers.12 Secondary glioblastomas are less frequent and have only been studied in comparatively small numbers.149,150 Such patients will generally have been treated by irradiation and/or chemotherapy. Glioblastomas show the greatest numbers of genetic abnormalities among the astrocytic tumors, and clear patterns of genetic aberrations are emerging. The TP53 gene is only mutated in about 37% of glioblastomas.60 However, the p53 pathway is targeted by mutations of other genes coding for proteins that control cellular p53 levels. The two genes whose products are involved in controlling p53 levels are p14(ARF) and MDM2. p14(ARF) controls the activity of MDM2,151 which in its turn controls the breakdown of p53.152 Loss of both copies of the p14(ARF) gene or amplification and overexpression of MDM2 will lead to the rapid breakdown of wild-type p53 protein, resulting in a cell with little or no wild-type p53 (see Fig. 1-6). The vast majority of glioblastomas (over 70%) have either no wild-type p53 or no p14(ARF) or have amplification and overexpression of MDM2 as mutually exclusive genetic abnormalities.60 Methylation of the p14(ARF) promoter with decreased or no expression are further mechanisms that have been shown to be involved in some tumors.153
Similarly, one or another of the genes coding for proteins involved in the control of entry into the S-phase of the cell cycle (the retinoblastoma pathway) is mutated in glioblastomas (see Fig. 1-6). At the beginning of the G1 phase of the cell cycle, RB1 is unphosphorylated. Unphosphorylated RB1 normally sequesters the E2F transcription factors.154 Entry into S-phase is initiated by the release of the E2F transcription factors by the newly phosphorylated Rb1 at the restriction point in G1. Loss of both wild-type copies of the RB1 gene, resulting in nonfunctional or absent RB1 proteins or inappropriately phosphorylated RB1, will result in any expressed E2F being free to initiate transcription of the genes necessary for entry into S-phase. Inappropriate phosphorylation may be achieved in glioblastomas with wild-type RB1 by either loss of wild-type p16 expression or overexpression of CDK4 (secondary to amplification of its gene). Both of the latter events would make inappropriate phosphorylation of wild-type RB1 more likely with the release of the E2Fs. p16 normally binds CDK4 and inhibits the formation of the CDK4/cyclin D1 heterodimer that phosphorylates RB1.155 In the absence of p16, all expressed CDK4 is available for heterodimer formation. When CDK4 is amplified and overexpressed in the presence of normal levels of p16, there will be excess CDK4 available for heterodimer formation. One or the other of these genetic abnormalities is present in over 70% of glioblastomas; they are, with very few exceptions, mutually exclusive.60 In addition, loss of RB1 expression due to promoter methylation has been described in glioblastomas.156 While disruption of both the p53 and Rb1 pathways seems essential for glioblastomas, the ways in which the pathways are rendered dysfunctional may confer slightly different biological characteristics on the individual tumors.
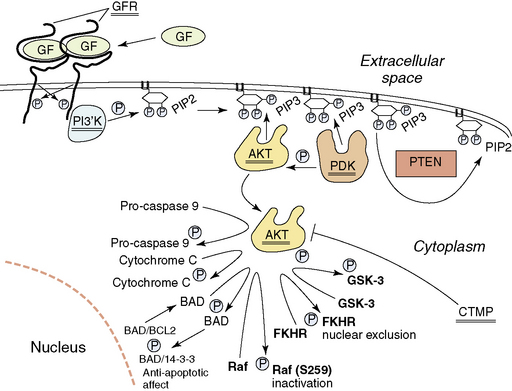
In addition to disruption of the p53 and Rb1 pathways, normal growth factor receptor signaling and signal transduction is also frequently disrupted in the glioblastomas. Starting at the surface of the cell, we will describe the abnormalities found (Fig. 1-7). About 35% of glioblastomas have amplification of the epidermal growth factor receptor (EGFR) gene (7p11–12). When amplified, this gene is always overexpressed, but it may also be overexpressed in the absence of amplification. Rearrangements of the amplified gene occur in almost half of the tumors with amplification. The most common rearrangement results in a transcript that is aberrantly spliced, but remains in frame157–159 and codes for a mutated EGFR that has lost 267 amino acids of its extracellular domain and does not bind ligand.160–161 This mutated EGFR is constitutively activated, and attempts are ongoing to target therapy to this aberrant cell surface molecule.162,163 Other rearrangements of the amplified EGFR gene occur less frequently. These may result in abnormalities of the cytoplasmic domain, but also lead to increased signaling by the aberrant receptors.164
Signal transduction from the activated EGFR is through the RAS-RAF-MAP kinase pathway and through the activation of the PI3K (phosphoinositide 3-kinase)/AKT pathway. Aberrations of the RAS-RAF-MAP kinase pathway are uncommon in glioblastomas. However, signaling through the PI3K/AKT pathway is frequently aberrant. Normally, activated growth factor receptors’ cytoplasmic domains activate the heterodimeric PI3K, which then phosphorylates local PIP2 (phosphatidylinositol-3,4-diphosphate) to PIP3 (phosphatidylinositol-3, 4,5-triphosphate) (Fig. 1-7), resulting in the recruitment of AKT and PDK1 (3′-phosphoinositide-dependent kinase-1) to the inside of the membrane, due to their binding to PIP3 through PH domains. Here AKT comes into contact with PDK1, which phosphorylates and activates AKT. Deregulation of this process occurs in a number of ways. Activating mutations of the PIK3CA (phosphotidylinositol 3-kinase, catalytic alpha) gene that codes for the catalytic component of the heterodimeric lipid kinase PI3K, resulting in constitutive activation, have been reported in WHO grade III and IV astrocytic tumors, some medulloblastomas and anaplastic oligodendrogliomas.165–167 This would result in inappropriate levels of PIP3. The levels of PIP3 are negatively controlled by the presence of the dual-specificity phosphatase PTEN protein.168–169 One of its major substrates is PIP3, which it dephosphorylates to PIP2.170 The PTEN gene is located on 10 q, and almost 90% of glioblastomas lose the region containing one PTEN gene at 10q23–24.171–173 The retained copy of PTEN is mutated in over 40% of glioblastomas; thus, these tumors lack wild-type PTEN and cannot dephosphorylate PIP3 to PIP2174 permitting stable high levels of PIP3 and activation of the Akt pathway (Fig. 1-7). This results in, among other things, decreased likelihood of apoptosis and the facilitation of HIF-1 (hypoxia inducible factor -1) activity.175 Amplification and overexpression of AKT have also been described in at least one glioblastoma176 and methylation and transcriptional downregulation of the CTMP gene (a negative regulator of AKT) have also been documented,177 further events that would activate the AKT pathway. This is supported by reports on the affect of Akt activation in animal models of astrocytoma.178 There is also data to suggest wild-type PTEN may contribute to growth arrest independently of the AKT pathway.179 A more detailed discussion of the pathways targeted in gliomas has recently been published.180
There have been some attempts to correlate the genetic findings to patient outcome following conventional therapy (debulking surgery followed by 50 + 60 Gy irradiation). Amplification of the EGFR gene has not been shown to uniformly result in a better or worse outcome.114,181–187 Combining the abnormalities has suggested that glioblastoma patients whose tumors have a disrupted RB1 pathway, disrupted p53 pathway, and no wild-type PTEN have the worst prognosis.188 Similar findings have been reported for anaplastic astrocytomas WHO grade III.189 While we still have no really well-documented genetic biomarkers for the astrocytic tumors that are diagnostic, predictive, prognostic, or therapeutic-response indicators (with the exception of methylation of the MGMT gene—see following discussion190), these insights into the genetic abnormalities of astrocytic tumors provide us with many potential therapeutic targets as well as an understanding of the complexity of the problem we are dealing with.
It is tempting to try to sort the genetic findings into a series of events explaining how de novo and secondary glioblastomas might develop. Both have disrupted p53 and Rb1 pathways—but these have occurred in different ways. In de novo tumors, amplification of the 12q14 region encompassing the CDK4 and MDM2 genes and resulting in their overexpression is found. This will disrupt both pathways by a single genetic event. Homozygous deletion of the region on 9 p encompassing the genes coding for p16 (CDKN2A), p15 (CDKN2B) and p14(ARF) (p14(ARF)) requires two genetic events (deletion of both alleles), and is also mainly found in de novo glioblastomas.116,191 Occasionally, de novo glioblastomas may also show greater numbers and more complex patterns of mutations with loss of one allele of each of TP53 and RB1 and mutation of the retained alleles, requiring 4 genetic mutational events. However, this is the rule in the secondary glioblastomas.116,191 The data from the diffuse astrocytomas WHO grade II and the anaplastic astrocytomas WHO grade III indicate that they have, in over 60% of cases, no wild-type TP53 gene through loss of one allele and mutation of the other.60 Glioblastomas derived from these tumors would be expected to have a similar pattern of TP53 mutations and this is what is found. The secondary glioblastomas also lose their functional Rb1 pathway but most often by a further two genetic events—loss of one allele and mutation of the retained RB1. In addition, losses on 10 q, encompassing the PTEN gene, occur in the majority of secondary glioblastomas, while EGFR amplification is uncommon.114 There are no data on the incidence of mutation of the retained allele of PTEN in secondary glioblastomas.
Methylation of tumor suppressor gene and DNA repair gene promoters has not received the same degree of attention as deletions and mutations, for practical methodological reasons. While some studies had shown methylation of known tumor suppressor genes, the association of MGMT (O(6)-methylguanine-DNA methyltransferase) methylation with improved survival in glioblastomas treated with alkylating agents made the field very relevant clinically.192 This was subsequently confirmed in a clinical trial of combined radiation and temozolomide treatment.123,190 The findings indicated that patients with methylation of the MGMT gene, and thus no expression or very limited expression of the protein, had a significant survival benefit—even with radiotherapy alone.190 The cytotoxic and mutagenic actions of temozolomide have been attributed to its ability to form DNA adducts by methylation at the O6 position of the purine base guanine to form O6methylguanine.193 Repair of these adducts is mediated by MGMT, which removes them from DNA in a single step mechanism, without cofactors but with the consumption of one MGMT molecule per adduct.194 Thus constant expression of the MGMT protein is necessary for maintenance of the repair process.
Besides the well-characterized genes described above, astrocytic tumor genome analysis has shown many relatively consistent copy number alterations in additional chromosomal regions. At least some of these regions are likely to harbor novel oncogenes or TSGs, or their break points may be involved in the formation of fusion oncogenes. Some of the recurrent chromosomal imbalances include loss of 1p36, 6q23–27, 9p21–24, 10, 13p11–13, 13q14–34, and 22, and gain of 7.120,195–197 The losses at 1p36 will be discussed in relation to the total 1 p deletions seen in oligodendrogliomas in the next section. The consequences and genes targeted by many of these changes have yet to be identified.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


