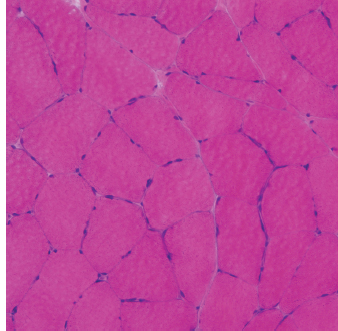
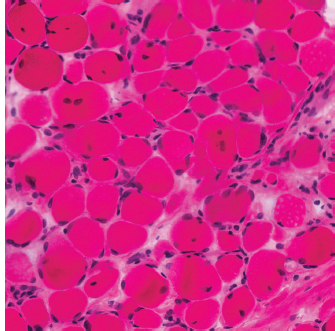
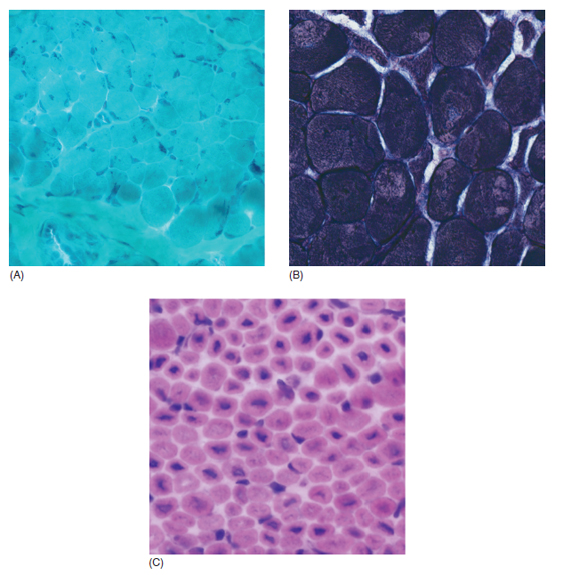
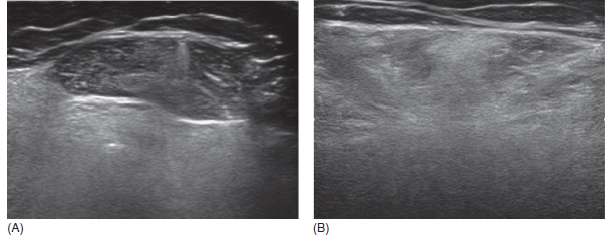
6 A traditional approach to diagnostic evaluation in neuromuscular disease centers on a thorough clinical history, physical examination, family history, and ancillary tests such as serum creatine kinase (CK) levels, neurophysiology, and muscle biopsy. The discovery of the DMD gene as the cause for Duchenne muscular dystrophy (DMD) in 1987 initiated the genetic era in neuromuscular disorders. The ever-increasing availability of genetic testing is challenging traditional approaches to diagnosis. Current paradigms for pediatric muscular dystrophies and myopathies continue to follow the traditional approach with respect to the clinical history, physical examination, and family history to narrow the differential diagnosis to one or a few disorders that can then be tested directly with a genetic test (1). For common disorders with distinctive phenotypes such as DMD, myotonic dystrophy, facioscapulohumeral dystrophy (FSHD), and spinal muscular atrophy (SMA), genetic testing has become a first-line test based on clinical history and examination. The development of new, rapid, inexpensive, and expansive sequencing technology, termed next-generation sequencing (NGS) is further challenging traditional approaches and early use of sequencing is becoming more accepted (2). As the role of genetic testing becomes more prominent, the role played by ancillary testing has diminished. In this chapter, we review the role of ancillary testing and genetics in the evaluation of patients with suspected neuromuscular disease and discuss the changing paradigms presented by emerging genetic technologies. Evaluation of a child with a suspected myopathy or muscular dystrophy starts with a thorough history and physical examination. Differential diagnosis is shaped by clinical features, age at onset, and a dominant, recessive, or X-linked inheritance pattern (3). A number of different classification and diagnostic schemes have been proposed (4,5). These algorithms are complicated, however, by the ever increasing number of overlapping disorders. Recognition of the relative frequency of disorders can guide the differential diagnosis by prioritizing more common diagnoses. DMD, FSHD, and myotonic dystrophy are substantially more common than any of the limb-girdle muscular dystrophies (LGMDs), congenital muscular dystrophies (CMDs), or congenital myopathy phenotypes in most populations (6). These more common diagnoses are often identifiable by clinical features, which prompt appropriate confirmatory genetic testing. Among CMDs, collagen VI–related muscular dystrophies (COL6-RDs) are most common except in Japanese populations where Fukuyama muscular dystrophy predominates due to a common founder mutation in the FKTN gene (7,8). COL6-RDs accounted for almost half of the genetically confirmed CMD patients in one study in the United Kingdom, with merosin-deficient CMD and alpha-dystroglycan-related CMD also relatively common, accounting for nearly 20% and 30% of the cases, respectively (8). RYR1-related central core myopathies are the most common congenital myopathies in the United Kingdom, accounting for half of the genetically confirmed cases, with SEPN1 and ACTA1 each accounting for 16% of the genetically confirmed congenital myopathies (9). In the newborn period, patients with neuromuscular disorders typically present with muscle weakness, hypotonia, and joint contractures. Discrimination of neuromuscular vs. nonneuromuscular etiology in the hypotonic infant can be complex; however, infants with encephalopathy, seizure, or abnormal movements are suggestive of a central rather than peripheral etiology (10,11). Respiratory and feeding difficulties are common in both neuromuscular and nonneuromuscular disorders. A history of polyhydramnios and decreased fetal movements is common during pregnancy in patients with neuromuscular diseases. Normal cognitive functioning in the newborn period supports a neuromuscular diagnosis. Since it is common, evaluation of any hypotonic infant should include the consideration of congenital myotonic dystrophy. Examination of the mother for clinical features of myotonic dystrophy may assist in making a diagnosis in the child. It should be noted that the mother is often unaware of her diagnosis until the birth of an affected child. Clinical features can assist in narrowing a differential diagnosis. Centronuclear myopathy or nemaline myopathy patients often have profound facial weakness in the neonatal period particularly in the lower face (5). Ptosis and ophthalmoparesis are also common features seen in severe congenital myopathies in the newborn period. Distal hyperlaxity and abnormal scar formation suggest a COL6-RD. Scoliosis and spinal rigidity are prominent features in myopathies related to SEPN1, LAMA2, LMNA, collagen VI, and RYR1. Cardiac involvement can be a clue to guide the differential diagnosis. Cardiac involvement is seen in some cases of TTN- and MYH7-related myopathies but is generally uncommon in most congenital myopathies (12,13). Cardiac findings are common in DMD, Emery-Dreifuss muscular dystrophy (EDMD), and myotonic dystrophy, but onset is usually in adolescence or adulthood, despite the onset of skeletal muscle weakness much earlier. Among CMD patients, FKRP- and FKTN-related muscular dystrophies are most commonly associated with cardiac problems. Serum CK is often the first test performed for suspected neuromuscular disorder and can lead directly to specific genetic testing in the setting of typical presentations of common disorders, such as DMD. CK is an enzyme with wide expression in many tissues but is enriched in tissues specialized for rapid energy metabolism such as skeletal and cardiac muscle. An elevation in CK suggests disruption of the permeability of the sarcoplasmic membrane, allowing CK to leak from the cytoplasm. Disruption of the sarcoplasmic membrane is common in dystrophic disorders associated with mutations in the dystroglycan-associated complex (DAC) such as dystrophin, alpha-dystroglycan, merosin, and the sarcoglycans. In DMD, CK elevations are often 10 to 100 times the upper limit of normal (ULN), and combined with a typical clinical presentation, leave little doubt of a neuromuscular diagnosis. CK elevations are present even in presymptomatic boys with DMD. Newborn screening based on CK elevations has been proposed to facilitate early diagnosis. In one large-scale trial, 37,000 newborns were screened for DMD based on elevated CK levels in routine blood spots. Six DMD cases and three LGMD cases were identified based on CK levels greater than 2,000 U/L (14). While newborn screening has not been systematically adopted, the development of gene-specific therapies such as nonsense read-through and exon skipping suggests the importance of early identification of these patients. In the absence of newborn screening, the first symptoms in patients with DMD can be identified as young as 2 years of age; however diagnosis is delayed until a mean of 4.7 years, a diagnostic delay that is unchanged in 25 years (15,16). Simple changes in practice such as early evaluation of the CK in patients with gross motor delay decreases the time to diagnosis in these patients. CK elevation in asymptomatic and paucisymptomatic individuals is more complicated. Variations between individuals based on gender, race, and age are well established (17). Many factors unrelated to neuromuscular disease can cause elevations in CK (18). Systemic causes for CK elevation include viral illness, trauma, physical exertion, and endocrine disorders (especially thyroid and parathyroid). Medications such as HMG-CoA reductase inhibitors (statins) are a common cause of elevated CK. Other common medications that can cause CK elevation include fibrates, antiretrovirals, beta-blockers, and isotretinoin. Further, CK can be elevated as a secondary factor in patients with neurogenic disorders such as SMA or amyotrophic lateral sclerosis (ALS), particularly during periods of rapid denervation. While reported normal ranges vary between laboratories, current evidence-based data suggests ULN of 97.5th percentile for serum CK. Patients with CK elevations greater than 1.5 times ULN based on age and race should be evaluated for a possible neuromuscular disorder (325 U/L for non-black females, 504 U/L for non-black males, 621 U/L for black females, and 1201 U/L for black males) (19). Clinical neurophysiology or electrodiagnosis is often obtained early in the evaluation of children with suspected neuromuscular disorders and is considered an extension of the physical examination. Electrophysiology consists of three individual studies, nerve conduction studies (NCSs) assessing the integrity of sensory and motor nerves, repetitive stimulation assessing the integrity of the neuromuscular junction, and routine needle electromyography (EMG) to assess the integrity of muscle architecture. Technical aspects of these tests are detailed elsewhere in this volume and are especially important to consider when NCSs/EMGs are performed in infants (20). Of note, peripheral nerves structurally mature with age, with an increase in the number of large fibers from birth to 8 years and complete myelination by 5 years. Thus, nerve conduction values for amplitude and conduction velocities must be adjusted for age (21). Routine needle EMGs/NCSs are associated with a degree of discomfort, and studies in children are usually limited to the extent of muscles studied, with the selection of those most likely to be diagnostic (proximal muscles in suspected myopathic disorders). The use of disposable smaller “facial electrodes” ensures a sharp point and small needle diameter. Topical anesthetic or light sedation can facilitate studies in poorly cooperative children. Routine studies require active contraction of muscles, and graded voluntary muscle activation is challenging in the pediatric population. Measurement of neuromuscular transmission jitter can be performed without patient cooperation by electrical stimulation of nerve branches (22). Repetitive nerve stimulation is usually performed when congenital myasthenia gravis is suspected. A more sensitive measurement of neuromuscular junction transmission is by the measurement of jitter by single-fiber EMG and its variations (23). Children with low tone, weakness, gait abnormalities, or other symptoms and signs typical of neuromuscular disease are often referred for EMG/NCS as a first step in the evaluation. EMG/NCS primarily separates peripheral neuropathies or motor neuropathies from primary muscle disorders. In one review of 122 children under the age of 3 years with hypotonia and weakness, NCS and EMG were supportive of SMA in 48%, CMD in 16%, and neuropathy in 8% (24). In another large case series, 39% of the cases referred for pediatric EMG/NCS showed neurogenic findings and were ultimately diagnosed as primary neurogenic disorders (25). In that same series, muscle disorders were ultimately diagnosed in 10% of the cases and included congenital myopathies, CMDs, DMD, and others. A central nervous system (CNS) disorder was diagnosed in 26%, and 22% ultimately lacked a neurologic diagnosis. It should be noted that in many cases of myopathy, EMG findings may be normal or show neurogenic changes. In one series, mild-to-moderate neurogenic changes were the only EMG findings in 36% of the patients ultimately diagnosed with a myopathy (26). For patients with suspected myopathic disorders, the utility of the EMG/NCS is primarily to exclude neuropathic conditions, provide objective evidence of myopathy, characterize the distribution (distal vs. proximal, and symmetry), and identify targets for muscle biopsy (27). While EMG findings can confirm the presence of a myopathic disorder, EMG cannot determine underlying pathologic processes, and a muscle biopsy or genetic testing is necessary to make a specific diagnosis, which is altered in many myopathies, by documenting changes in motor unit action potential waveforms. Abnormal spontaneous discharges include fibrillation potentials and positive sharp waves. These occur in both neuropathic and myopathic disorders and thus are not discriminating. The presence of myotonic discharges is traditionally associated with myotonic disorders (myotonic dystrophies, myotonia and paramyotonia congenital, and channelopathies), but they were also encountered in other forms of muscle disease (28). A 12-year review of 2,234 children revealed that 11 had myotonic discharges without other abnormalities on EMG that contributed to the diagnosis of myotonia congenital, paramyotonia congenital, congenital myopathy, and Pompe disease (29). Another group of eight patients had both myotonic discharges and myopathic motor units, which led to the diagnoses of congenital myopathy and non-Pompe glycogen storage diseases. Characteristic findings in myopathic disorders include short-duration, polyphasic, and low-amplitude motor unit potentials (22). It is important to note that interpretation of motor unit waveforms in children younger than 2 years requires experience because muscle fibers are of lesser diameter, and normal motor units may appear myopathic (30). Fibrillation potentials, positive sharp waves, and myopathic motor units are observed in most muscular dystrophies and myopathies including LGMD, EDMD, FSHD, myotonic dystrophy, congenital myopathy, and, inflammatory myopathies (31). While NCSs are usually normal in primary muscle disease, some disorders such as merosin-deficient CMD may show slow motor conduction velocities without signs of denervation (32–34). Further, there may be a progression of motor nerve slowing over time in an individual patient (35). With myotonic dystrophy type 1, there is a mild and mostly subclinical neuropathy (36). EMG can supplement other studies such as muscle biopsy. Among 17 children undergoing surgical procedures for the repair of clubfoot deformities without preoperative diagnoses, EMG combined with muscle biopsies at the time of surgery were helpful showing 70% with myopathic features (37). Data from a Mayo Clinic review of 72 children who had both an EMG study and a muscle biopsy showed that EMG was 91% sensitive and 67% specific in identifying myopathic disorders (28). Forty-six percent had an abnormal EMG study and a myopathic disorder (biopsy or genetic testing); 17% had an abnormal EMG but no pathologic or genetic testing evidence of a myopathic disorder; 4% had a normal EMG but a metabolic myopathic disorder; and 36% had normal studies. Congenital myopathies were the most commonly identified disorders in this review. All had abnormal EMG studies. While myotonic discharges are readily identified in adults with myotonic dystrophy, EMG findings in infants with congenital myotonic dystrophy may be absent months to years after birth, and the discharges may be sparse and atypical (ie, of shorter duration) (31). Electrical impedance myography (EIM) is a noninvasive and painless procedure that assesses changes in muscle structure by passing low-intensity alternating current through one set of surface electrodes and recording the voltages with another set of surface electrodes (38). Changes in muscle composition including atrophy, edema, and fibrosis can be identified with EIM since these changes alter the underlying conductance of the muscle tissue. EIM has been used to characterize SMA and can successfully differentiate types 2 and 3 SMA (39). Furthermore, EIM has been proposed as a method to grade the severity and follow the progression of muscle disease and may become an important outcome for clinical trials. In this respect, EIM has been shown to distinguish boys with DMD from healthy subjects, and EIM measurements correlated with functional outcomes in boys with DMD (40). A muscle biopsy is warranted in a child with weakness when CK elevation and/or myopathic features on EMG, imaging, or physical examination are present. Historically, most muscular dystrophies and myopathies were defined by features on the muscle biopsy, and muscle biopsy has been the mainstay of diagnostic testing (41). Muscle biopsy can be diagnostic of a specific disorder in many cases, including DMD, merosin-deficient CMD, sarcoglycanopathies, and dystroglycanopathies. Congenital myopathies such as central core myopathy, nemaline myopathy, and central nuclear myopathies are defined primarily by their histologic appearance and were considered distinct clinical entities until the identification of the genes underlying these disorders. Significant heterogeneity within myopathy subtypes and overlap between them has blurred the distinctions between disorders that were once thought to be distinct. Selection of the muscle for biopsy should be guided by the distribution of weakness. A moderately affected muscle is preferred since very weak muscle shows severe changes that obliterate specific findings. Muscle imaging by ultrasound or MRI can direct selection of the biopsy site and avoid sampling of muscle that is either too severely affected or relatively unaffected (42). Muscle biopsy can be obtained through an open incision (43) or using a specialized needle (44). Needle biopsy procedures have the advantage of being less invasive and leaving a smaller scar but has the disadvantage of limited visualization of the sampling site and technical difficulty in very young children. Muscle biopsy samples should be taken immediately to the processing laboratory on saline-moistened gauze. Immersion of the sample or attempts to wash the sample can result in significant tissue artifacts. Once in the laboratory, samples are split, with half fixed in formalin and half frozen to preserve enzymatic activity. Detailed handling procedures and utility of the different stains are outside the scope of this chapter but are well reviewed in the literature (45,46). Hematoxylin and eosin (H&E) and modified Gomori trichrome stains are routinely performed to evaluate overall tissue architecture and organization. Adenosine triphosphatase (ATPase) stains allow distinction of type 1 and type 2 fibers. Nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), succinic dehydrogenase (SDH), and cytochrome oxidase (COX) are used to evaluate enzyme activity and distribution within the muscle fibers. Additional immunohistochemical stains are selected depending on the clinical question and include stains for sarcolemmal proteins such as dystrophin, sarcoglycans, dysferlin, caveolin, merosin, and others. Normal muscle structure shows polygonal muscle fibers with peripherally located nuclei. Fibers are uniform in size and shape and surrounded by a thin layer of connective tissue (endomysium) (Figure 6.1). It should be noted that fiber size is dependent on age. An average fiber size of 15 µm is expected in neonates, gradually increasing to 50 µm in adolescent children and adults (41). Muscle biopsies in neonates are often deferred until after 6 months of age due to difficulty in interpretation because of the small fiber size. Discrete bundles of muscle fibers (fascicles) are surrounded by a connective tissue layer, the perimysium, which also contains nerve fibers and blood and lymphatic vessels. Fascicles are bundled together by a thick connective tissue layer, the epimysium, which is contiguous with the tendon sheath. FIGURE 6.1 Normal muscle demonstrating polygonal fibers of uniform size with peripherally placed nuclei (hematoxylin and eosin [H&E] stain). Histopathology in muscular dystrophies is generally distinct from myopathies by the prominence of dystrophic features such as fibrosis, fatty infiltration, and necrosis (Figure 6.2); however, severe, early-onset congenital myopathies may also have a dystrophic appearance (47,48). Myopathic features commonly seen in muscle biopsies include central displacement of nuclei, atrophy or hypertrophy of fibers, degenerating/regenerating fibers, fibrosis, and fatty replacement. These features are somewhat nonspecific and can be seen in many different myopathic disorders. Congenital myopathies may have specific findings such as central cores, nemaline rods, or central nuclei (Figure 6.3). In many cases, congenital myopathies are defined by their histologic appearance, but genetic discoveries continue to blur distinctions between them. Specific histopathologic features are suggestive of more targeted diagnoses and can direct the selection of genetic tests. FIGURE 6.2 Dystrophic muscle biopsy from a 4-year-old boy with DMD. Note the marked variation in fiber size, hypertrophic fibers, necrotic fibers, and fibrosis (hematoxylin and eosin [H<E] stain). FIGURE 6.3 Muscle biopsy section from a patient with (A) nemaline myopathy demonstrating prominent cytoplasmic inclusions on Gomori trichrome stain, (B) central core myopathy demonstrating areas of central clearing in NADH-TR stain, and (C) centronuclear (myotubular) myopathy demonstrating immature fibers with predominantly centrally placed nuclei on hematoxylin and eosin (H&E) stain. Muscle imaging is an increasingly important tool in the evaluation of patients with suspected neuromuscular disorders. Imaging techniques including ultrasound (US), MRI, and CT have the advantage over neurophysiology and biopsy of being noninvasive and readily available at most centers. The use of muscle imaging in clinical settings is largely focused on the targeting of muscle biopsy and pattern recognition in the guidance of differential diagnosis. Research applications are advancing rapidly and are focused on the use of imaging markers of disease progression. In this regard, development of quantitative techniques has been an especially important area of investigation. Muscle US was first introduced in the 1980s and was promoted as a tool for a more accurate selection of muscle biopsy site (42,49,50). In recent years, US has been used increasingly as a tool for pattern recognition to aid in differential diagnosis (51). Normal muscle has low echodensity resulting in a black appearance on US. Underlying connective tissue appears bright, outlining the epimysial and perimysial architecture. In longitudinal orientation, the pennation pattern of individual muscles can be identified, and in the transverse plane, the muscle has a more speckled appearance. Bone, subcutaneous fat, and vascular structures can also be easily detected. Echodensity increases with age as muscle is replaced by fibrous connective tissue and fat. In patients with suspected neuromuscular disorders, US can be used to assess both echogenicity (degree of fibrosis, fatty replacement, and overall degeneration of the muscle architecture) and size (atrophy/hypertrophy) of muscle. Specific features can be identified in some disorders, including a “central cloud” phenomenon in the rectus femoris in patients with COL6-RD and marked sparing of the rectus femoris in RYR1-related myopathies (Figure 6.4) (52,53). Evaluation of deeper muscles in patients with severe disease may not be possible with US due to echogenicity of more superficial muscles. In very severely affected muscle, the underlying bone echo, which is very prominent in normal muscle US, can be completely obliterated. Prominence of the bone shadow has been used as a marker for grading the echodensity of the overlying muscle (50). FIGURE 6.4 Ultrasound image of rectus femoris in patients with (A) RYR1-associated myopathy and (B) collagen VI–related muscular dystrophy (COL6-RD). Note the sparing of rectus femoris and marked increase in echogenicity in other muscle groups in the patient with RYR1-associated myopathy. In COL6-RD, the rectus is affected centrally and spared peripherally. The resulting “central cloud” phenomenon is pathognomic of COL6-RD. In both cases, the normal bone echo is obscured by the involved muscle. Source: Image courtesy of Jahannaz Dastgir, MD, Columbia University. US has the advantage of portability, low cost, and immediate availability in the clinic setting. Serial evaluations can noninvasively identify progression. However interpretation is highly dependent on the skill of the examiner. In addition, technical differences related to equipment and different system settings complicate interrater reliability. US has the unique advantage of being a dynamic study, allowing the examiner to visualize movements in the muscle including fasciculations (54). CT of muscle can be performed rapidly and can detect changes in muscle size and volume and structure. Myopathic changes in muscle tissue are characterized by fibrosis and fatty replacement and are easily identifiable on CT as areas of low attenuation. CT, however, has poor soft-tissue contrast to differentiate inflammatory changes (edema) (55). The main disadvantage of CT is the radiation exposure, which has led to a decline in its routine use (55). MRI has largely replaced CT since it offers significantly better spatial resolution and requires no radiation exposure. While necessary sequences for MRI take longer to obtain compared with CT, most can be completed in a relatively short time frame. Typical MRI protocols for muscle imaging include T1-weighted, T2-weighted, and fat-suppressed (short tau inversion recovery [STIR]) images through hips, thighs, and calves. In most cases these studies can be done with little or no sedation in 15 to 30 minutes in children over 5 years old; however, sedation may be required for imaging in younger children. Normal muscle is intermediate in signal intensity on all sequences. Evidence of muscle disease such as fatty replacement of muscle and fibrosis can be identified easily on T1- and T2-weighted scans as areas of increased signal. Edema, which can precede these more chronic changes, can be distinguished from fibrosis and fatty infiltration on fat-suppressed, STIR images as areas of increased signal. Standardized rating scales have been proposed but are not widely used clinically (56–58). Quantitative measures of fatty infiltration and fibrosis are active areas of investigation with an interest in developing reliable indicators of progression, but these are not used routinely in clinical setting (59–61). From an imaging perspective, muscle can change in a limited number of ways (mass, shape, and signal intensity). Chronic myopathy results in fatty replacement without atrophy, while denervation results in atrophy. Since muscle changes in MRI are limited, a pattern recognition approach combining imaging characteristics and clinical features is most helpful. Changes in muscle can be described in four basic areas (62): (a) distribution and symmetry—focal vs. diffuse involvement, (b) morphology—size (atrophy, hypertrophy, pseudohypertrophy) and shape of muscle, (c) T1 changes—fatty replacement, usually diffuse in later stages and patchy in early stages of disease, and (d) T2 changes—largely due to edema suggesting inflammation. MRI of muscle in DMD and Becker muscular dystrophy (BMD) patients shows prominent involvement in the proximal leg, long head of biceps femoris, vastus lateralis, and rectus femoris. Biceps and gluteus maximus are less involved and progress more slowly over time (68). In the lower leg, there is greater involvement of posterior muscles (gastrocnemius greater than soleus) than anterior compartment muscles (anterior/posterior tibialis). Severity of MRI findings in DMD patients has been correlated with the severity and degree of fibroadipose tissue replacement in biopsy samples from extensor digitorum brevis muscle (69). MRI findings in BMD have been described and include prominent involvement of gluteal muscles, adductor magnus, long head of biceps femoris, semimembranosus, and the vasti; however, this pattern was not sufficiently distinct to clinically separate BMD from LGMD (70). Specific patterns have been identified for some CMD and congenital myopathy phenotypes. In some cases, imaging findings can directly inform confirmatory genetic testing and obviate the need for muscle biopsy. Patients with myopathies due to RYR1 mutations include a broad clinical spectrum from severely affected infants to mildly affected adults. Histopathologic findings are diverse and can include central core disease, multi-minicore disease, congenital fiber-type disproportion, and others. In contrast to the variability in clinical phenotypes, imaging characteristics are similar in most patients regardless of clinical and histopathologic findings (71). Imaging features include prominent involvement of the gluteal muscles, adductor magnus, sartorius, and vastus interomediolateralis and the sparing of rectus femoris and adductor longus (Figure 6.5) (72,73). These findings are easily identifiable by both US and MRI. There is some overlap with SEPN1-related myopathies, given severe involvement of the sartorius compared to gracilis, but sparing the posterior thigh muscles and vasti differentiates them (64). Patients with susceptibility to malignant hyperthermia due to RYR1 mutations in the absence of other clinical features have normal imaging (73). Atrophy of the sartorius with preservation of the rectus femoris is the hallmark of rigid spine muscular dystrophy type 1 (RSMD1) due to SEPN1 mutations (73). Posterior thigh muscles and adductor magnus are also involved while other muscle groups are spared (64). Lower leg muscles are less severely affected, and on whole-body MRI, sternocleidomastoid muscles are severely affected, which may be a pathognomonic feature (66). In milder cases with SEPN1 mutations, the sartorius may be the only significantly involved muscle by imaging features and may be difficult to identify due to severe atrophy. Overlapping clinical disorders such as CMD due to LMNA mutations are distinguished from SEPN1-related disorders by sparing of the sartorius. FIGURE 6.5 Transverse T1-weighted image through the thigh of a patient with RYR1-associated myopathy demonstrating sparing of the rectus femoris. Source: Image courtesy of Jahannaz Dastgir, MD, Columbia University. Patients with COL6-RD such as Bethlem myopathy (BM) and Ullrich congenital muscular dystrophy (UCMD) show diffuse involvement of posterior and lateral thigh muscles with relative sparing of the sartorius, gracilis and adductor longus. COL6-RD patients have a characteristic rim of abnormal signal in the vastus lateralis and gastrocnemius with sparing of the central areas (52,74). Involvement of the rectus femoris is prominent centrally (Figure 6.6). This phenomenon, termed central shadow or central cloud in the rectus femoris is pathognomonic in COL6-RD patients and is identifiable on all imaging modalities. In nemaline myopathy patients with mutation in NEB, rectus femoris is the most affected, with significant involvement of the tibialis anterior (63). Involvement of the tibialis anterior may be out of proportion to clinical involvement (66). Imaging characteristics of other congenital myopathy and muscular dystrophy phenotypes are less well characterized. Patients with ACTA1 mutations have broad clinical phenotypes and no specific imaging pattern; however, involvement of the lower leg is less significant than that seen in patients with NEB mutations. EDMD due to a mutation in emerin is distinguished from EDMD due to a mutation in lamin A/C by disproportionate involvement of the medial head of gastrocnemius (75). FIGURE 6.6 Transverse T1-weighted image through the thigh of a patient with Ullrich congenital muscular dystrophy demonstrating characteristic “central cloud” in the rectus femoris and rimming of vastus lateralis. Note also relative sparing of the sartorius and gracilis. Source: Image courtesy of Jahannaz Dastgir, MD, Columbia University. LGMD is a genetically and clinically heterogeneous group of disorders characterized by weakness in the shoulder and hip flexors. Patterns of specific muscle involvement in LGMD identifiable by muscle imaging have been suggested (55,73); however, clinical, histological, and imaging findings are largely overlapping. Imaging characteristics are most distinctive early in the course of disease before motor impairment becomes severe (65). Patients with LGMD2A due to mutations in CAPN3 show early involvement of gluteal, posterior/medial thigh, adductors, and semimembranosus. With clinical progression, involvement is more diffuse but the vastus lateralis, sartorius, and gracilis are spared to some extent (76). LGMD2B due to DYSF mutations show a more diffuse pattern than LGMD2A. Patients with DYSF mutations can have either LGMD or a distal myopathy phenotype, Myoshi myopathy. In both cases, muscle involvement is diffuse, but the calf muscles are the earliest and most severely affected (73). LGMD2I due to mutations in FKRP is the most common LGMD in North American and European populations. Muscle involvement in the thigh is reminiscent of that seen in LGMD2A, but selective involvement of the medial gastrocnemius and soleus in the lower leg is seen in LGMD2A but is absent in LGMD2I (77). In contrast, sarcoglycanopathy (LGMD2C-2F) and dystrophinopathy patients showed early involvement of the anterior thigh and little involvement of the lower leg, differentiating them from other LGMDs (55,73). Genetic testing in neuromuscular disease began with the discovery of mutations in the DMD gene as the cause for DMD in 1987 (78). Since then, the number of genes associated with neuromuscular phenotypes has grown rapidly. A comprehensive list of neuromuscular genes and phenotypes is maintained in the GeneTable of Neuromuscular Disorders (www.musclegenetable.fr) (79). The 2014 version of the GeneTable includes 685 different neuromuscular phenotypes associated with 360 different genes. New genes are identified regularly, with 27 new genes added in 2014. Application of genetic discoveries in the clinical setting was slow at first, but with the accelerating discovery of new genes and rapid improvement of the sequencing technology, genetic diagnosis has become an important part of the workup for any patient presenting with neuromuscular weakness. Funding for genetic testing varies by country and by insurance coverage. Third-party payers and even many physicians have been hesitant to pursue genetic testing since specific treatments for most neuromuscular disorders are not available. Coverage often is dependent on a demonstration of clinical utility. Given the increasing complexity of genetic testing, inclusion of a genetic counselor early in the diagnostic evaluation can often facilitate education for families, insurance preauthorization, and identification of appropriate genetic tests. In most cases, genetic testing is sent to specialized laboratories. Laboratories vary widely in cost and quality, even for the same test, and care must be taken to select both the lab and the genetic test appropriately. Sequencing panels are emerging using both traditional and NGS approaches. These panels can be helpful for disorders with significant heterogeneity but can be wasteful if clinical and diagnostic features suggest a more specific diagnosis. Selection of the appropriate genetic test and appropriate laboratory for testing can be facilitated by a number of online resources including the Genetic Testing Registry at the National Institute of Health and www.genetests.org. From a diagnostic standpoint, genetic testing offers a definitive diagnosis. The clinical utility of a definitive diagnosis for patients and families in the midst of a prolonged diagnostic odyssey is difficult to understate. An accurate genetic diagnosis facilitates genetic counseling and estimation of recurrence risk, guides appropriate surveillance, and avoids unnecessary and costly testing. Clinical utility is demonstrated by a significant impact on both survival and quality of life for patients with disease-specific surveillance facilitated by accurate genetic diagnosis (80,81). Furthermore, development of specific treatments depends on having well-characterized, genetically distinct groups of participants. Participation in most clinical trials requires a genetic diagnosis. The benefits of genetic testing must be balanced with the costs, which can be high, especially if multiple genetic tests are required. The rapid evolution of NGS technologies and rapid drop in cost of sequencing will ultimately change this balance in favor of early genetic testing. As early as the year 2000, genetic testing was recommended as a first-line test, before either EMG/NCS or muscle biopsy in cases of suspected DMD/BMD, EDMD, FSHD, myotonic dystrophy, and congenital myotonias (31). Standards of care have been developed in recent years for DMD, congenital myopathies, CMDs, myotonic dystrophy, and others. Appropriate application of these standards depends on accurate and specific genetic diagnosis, even when a diagnosis is already made by biopsy or other means (80,82–84). Genetic testing as a first-line test is the current standard for DMD, myotonic dystrophy type 1, and SMA in patients with typical clinical presentations (82–85). Selection of genetic tests may depend on regional factors. In Japan, Fukuyama-type CMD (FCMD) is the most common CMD, accounting for almost half of the cases (7,87,88). The high number of FCMD patients in Japanese populations has been traced to the insertion of a 3-kb retrotransposon in the FKTN gene in a founder 102 generations ago (88). The high prevalence of this founder mutation in Japanese populations (1/188 individuals) suggests that molecular testing should be performed early in the evaluation of CMD patients in Japanese populations but should be a later consideration in other populations where FKTN mutations are more rare. Genetic testing is complicated by the large size of many of the genes involved in neuromuscular disorders. These include some of the largest genes in the human genome including TTN, DMD, NEB, and RYR1. Sequencing of these genes by traditional methods (Sanger sequencing) is time-consuming and very expensive. Initial efforts at molecular diagnosis identified mutational hot spots where disease-causing mutations clustered, ie, exons 44 to 51 in the DMD gene for DMD and the C-terminal transmembrane domain of the RYR1 gene for central core myopathy (78,89). To save costs, many clinically available tests for these large genes screen only mutational hot spots, and failure to identify a mutation does not rule out a disorder associated with one of these genes. Technological improvements have made comprehensive analysis of these genes possible and have led to the identification of significant mutations throughout these very large genes (90–92). Titin, encoded by the TTN gene, the largest gene in the human genome, encompassing 364 exons in a 100-kb-long transcript was first associated with tibial muscular dystrophy, due to mutations in the M-line domain (93). For some time, sequencing of the TTN gene focused primarily on this region of the gene. With newer technologies that allow sequencing of the entire gene, a variety of skeletal muscle and cardiac phenotypes have been associated with mutations throughout the TTN gene, but no clear genotype-phenotype association has emerged (92). In many cases, patients with suspected neuromuscular disorders have few distinguishing features and a large number of potential genetic etiologies. Genetic diagnosis in these cases is complicated by high levels of clinical and genetic heterogeneity. Rather than simplifying the diagnostic approach, the rapid discovery of new genes in these disorders has only added to the known heterogeneity and blurred what appeared to be specific genetic associations in early genetic studies. Among the genes causing LGMD phenotypes, for example, the 2014 GeneTable lists 8 autosomal dominant and 21 autosomal recessive genes. A dominant, recessive, or X-linked family history may guide a differential; however, in some cases such as COL6-RD, inheritance may follow a dominant or recessive pattern depending on the specific mutation (94). A striking example of the phenotypic variability from a single gene comes from mutations in the nuclear envelope protein lamin A/C encoded by the LMNA gene (OMIM 150330). Mutations in LMNA cause a diverse set of neuromuscular and nonneuromuscular phenotypes including EDMD, LGMD1B, a severe CMD with dropped head, Charcot-Marie-Tooth type 2B1 (CMT2B1), dilated cardiomyopathy 1A (CMD1A), familial partial lipodystrophy, Dunnigan type 2 (FPLD2), and Hutchinson-Gilford progeria syndrome (100). In turn, EDMD, a fairly specific clinical entity characterized by progressive weakness in a scapulo-peroneal-humeral distribution, early contractures in ankles and elbows, and cardiomyopathy can be caused by as many as three different genes, EMD, FHL1, and LMNA (101). In each case, the clinical phenotype is identical. Family history can help to narrow the search for the causative gene, but inheritance can be difficult to assess in small families and may not be completely informative since both EMD and FHL1 are X-linked and LMNA can be either autosomal dominant or recessive. Soon after the identification of the DMD gene in 1987 (78), clinical genetic tests quickly became available; however, this testing was technically challenging and costly. Due to the large size of the DMD gene, clinically available tests did not include a comprehensive analysis of the entire gene but focused on mutational hot spots where deletions were common. With the development of newer sequencing strategies, genetic diagnosis for DMD became widely available from the early 2000s (90). As genetic testing has become more readily available in the clinic, genetic testing for DMD has become a standard part of the workup for most cases. For DMD, the current standard for genetic testing consists of stepwise analysis for deletion/duplication mutations followed by direct sequencing of all 79 exons in cases where a deletion or duplication is not identified (84). Genetic testing is usually sent early in the evaluation of boys with appropriate clinical presentation and elevation in CK and obviates the need for muscle biopsy or other diagnostic tests. Using current technology, a genetic diagnosis can be made in more than 95% of the cases where clinical history and elevated CK suggest a diagnosis of DMD (90,102). Deletion or duplication mutations in the DMD gene are the most common and are identified in 72% of the cases, while nonsense mutations are identified in 17% patients (103). Generally speaking, BMD and DMD can be differentiated based on the reading frame rule. Nonsense mutations and deletions or duplications that disrupt the reading frame result in a DMD phenotype. Missense mutations and deletion or duplication mutations that leave the reading frame intact result in BMD. Mutations in the DMD gene are de novo in one-third of affected boys with the remaining two-third inheriting their mutation from their carrier mothers (106). Once a mutation has been identified in an affected boy, carrier testing for the mother is critical. With a high recurrence risk (50% for sons of carrier mothers), and many mothers still of childbearing age at the initial diagnosis, appropriate genetic counseling can give important guidance to family planning decisions. As with any genetic test, a normal finding on carrier testing does not exclude the possibility that an individual might pass the mutation to future offspring due to the possibility of germ-line mosaicism. In DMD, the risk that an individual with negative carrier testing has the mutation in the germ line has been estimated at 8.6%, resulting in a 4.3% chance that she will pass the mutant allele to future offspring (107). Technologies such as preimplantation diagnosis are available to families where carrier status has been confirmed. Daughters of carrier mothers are at 50% risk for carrier status and should be appropriately counseled about recurrence risk. Carrier testing should be offered when potential carriers are of appropriate age to consent to testing. It should also be noted that female carriers of DMD mutations can have a variety of manifestations of disease, including a full DMD phenotype (108). In most cases clinical features are mild and include myalgias and fatigability, but progressive cardiomyopathy has been described (109). Since carriers can have some manifestations of disease, especially cardiac, establishment of recurrence risk and identification of carrier status can inform medical decision making and surveillance for carriers (110). Diagnosis in the congenital myopathies has traditionally centered on findings in the muscle biopsy. Histopathologic classes including nemaline myopathies, core myopathies, centronuclear myopathies, and myopathies with congenital fiber-type disproportion have been well described (5). In each case, multiple genes have been identified with each histopathologic group (Table 6.1). Nemaline myopathy is caused by mutations in nebulin (NEB) in up to half of the cases and ACTA1 in 20% to 25% of the cases; however, as many as six other genes can cause a myopathy with nemaline rods (5). Since these disorders lack clinical or histopathologic features to differentiate them, genetic sequencing is necessary to make a specific diagnosis. Clinical features can sometimes narrow the differential diagnosis to a smaller group of genes. The presence of ophthalmoplegia in a patient with centronuclear myopathy on biopsy suggests a mutation in MTM1, RYR1, or DNM2. Cardiac involvement in a patient with otherwise nonspecific myopathy suggests a TTN or MYH7 mutation. Sequencing of many of these genes is complicated by the large size of the genes, which can make genetic testing cost-prohibitive. Nebulin is especially notable in this respect, with 183 exons. TABLE 6.1 Overview of Genes Associated With Congenital Myopathies CMD includes a group of disorders that vary widely in clinical features but share dystrophic features on muscle biopsy (Table 6.2). A combination of clinical features and ancillary testing such as muscle imaging and biopsy can direct specific genetic testing. COL6-RDs are the most common CMDs and present with distal joint hyperlaxity and proximal joint contractures. The presence of skin changes such as keloid scars and hyperkeratosis pilaris with distal laxity should prompt the consideration of a COL6-RD, even without biopsy or imaging tests. Patients with merosin-deficient CMD are not easily distinguished from other CMDs clinically but are readily identified by the absence of merosin on muscle immunohistochemistry. In these cases, genetic testing targeted to the LAMA 2 gene is readily available and confirms the diagnosis at a molecular level. The most genetically diverse group of CMDs is due to defects in the glycosylation of alpha-dystroglycan. These disorders include a spectrum from very severe in the Walker-Warburg and muscle-eye-brain phenotypes to the relatively mild LGMD phenotypes. While a disruption of glycoslylation of alpha-dystroglycan can be readily identified on muscle biopsy samples, distinction between the 12 or more genes identified to date including FKTN, POMT1, POMT2, FKRP, POMGNT1, ISPD, GTDC2, B3GNT1, GMPPB, LARGE, DPM1, DPM2, ALG13, B3GALNT2, and TMEM5 is only accomplished by sequencing each of the genes. TABLE 6.2 Overview of Genes Associated With Congenital Muscular Dystrophies Given the increasing complexity of genetic diagnosis including the increasing number of genes per phenotype and increasing numbers or phenotypes per gene and the inherent difficulty of sequencing many of the very large genes associated with neuromuscular disorders, selection of an appropriate genetic test can be very difficult. The dramatic expansion of sequencing capacity afforded by NGS techniques will likely result in a shift in traditional diagnostic paradigms, since many genes (or all genes) can be sequenced simultaneously at a relatively low cost. The Human Genome Project was launched in 1990, just 3 years after the discovery of the DMD gene, with a goal to sequence the entire human genome. Sequencing for the Human Genome Project started with traditional Sanger sequencing methods and ended by taking advantage of increasing computing power and high-throughput “parallel” sequencing techniques. Using these techniques, sequencing the first full human genome cost an estimated $300 million and involved the efforts of many hundreds of scientists. The completed sequence was published in 2004 (111). Taking advantage of lessons learned in sequencing the first human genome, new technologies emerged to greatly amplify the efficiency and parallelization of the sequencing. These advances in sequencing technology, broadly termed next-generation sequencing have allowed a dramatic decrease in the cost per base for sequence generation and has led to a rapid acceleration in the development of sequencing applications (112,113). A variety of technologies have emerged but all take advantage of the ability to rapidly sequence millions of short fragments of DNA (or cDNA) in a parallel fashion. Among the first to use NGS technologies, Lupski et al. identified a variant in the SH3TC2 gene in a family with CMT neuropathy by whole genome sequencing at an estimated cost of $50,000 per genome (114). Continued rapid development of these technologies has been promoted by the National Institutes of Health (NIH) and others with a goal toward providing a complete genome sequence for under $1,000 (115). Realization of these goals is anticipated to result in dramatic changes in the utilization of DNA sequencing in both clinical and research applications. While the issues of obtaining sequence data have been largely addressed, issues of data analysis including data storage, patient privacy, insurability, and handling of incidental findings remain significant (116–118). While NGS technology has made sequencing of the whole genome feasible, targeted NGS approaches have simplified some of the complex data analysis issues and allowed a more straightforward application for clinical use. Using this approach, samples of interest are subjected to an enrichment step during sample preparation that restricts the sequenced DNA to only those areas of interest. For exome sequencing, the targeted sequences are the exons of all genes. The exome sequence covers 30 million bases in 180,000 exons from 23,000 genes or about 1% of the total human sequence. Within that 1% of the genomic sequence is an estimated 85% of the pathogenic mutations. Thus exome sequencing can significantly limit the search space and at the same time identify mutations in almost every gene without a priori knowledge of the phenotype. A number of different exon capture technologies are available. All have gaps in coverage, and analysis of the resulting sequence data should acknowledge the possibility that some genes or exons may not be well covered (119). With current exome capture technologies, most leave 10% of exomes poorly covered and 3% of exons not targeted at all. Gaps in exome coverage are somewhat systematic and reflect difficulties in hybridization due to high guanine-cytosine (GC) content and other factors in the primary sequence. Rapid development in these areas is expected to improve coverage; however, gaps are likely to remain. Beyond exome sequencing, strategies have been developed to target different disorders by limiting the number of exons in the enrichment step to a relatively small number of genes of interest. Targeted gene panels such as this take advantage of the capacity of the sequencing technology to cover a large number of genes and yet limit the search for mutations to those genes with a relationship to the phenotype of interest (120). In one study, 267 genes selected from the Muscle GeneTable were selected for sequencing in eight myopathy patients with known molecular diagnoses and eight patients with neuromuscular disorders but without a molecular diagnosis (121). Pathogenic mutations were identified in 13 of the 16 patients with a variety of neuromuscular disorders. For disorders where genetic and phenotypic heterogeneity is the rule, genetic testing using panels of neuromuscular genes will likely change the diagnostic approach. This approach is likely to include genetic testing at a much earlier point, perhaps even before the use of ancillary tests such as EMG/NCS, imaging, and muscle biopsy, especially as costs for sequencing by exome or by targeted sequencing drop below those of traditional tests. Clinical use of targeted sequencing panels is rapidly evolving and targeted NGS panels for neuromuscular disorders are currently available from a number of commercial labs. Selection of the appropriate panel requires careful review of the genes included on the panel and assessment of clinical phenotype. A panel for 8 to 10 genes associated with nemaline myopathy, for example, may be sufficient if nemaline rods are identified on muscle biopsy. However, if the clinical phenotype and ancillary tests are less specific, a larger, more diverse panel may be more appropriate. Despite its enormous potential, there are pitfalls in using NGS. Sequencing using these technologies is inherently less precise on a per-base level than traditional Sanger sequencing. Analytical tools such as those to assess the quality of the base calls and repeated sequencing of the same base in different reads (read depth) can help to overcome this imprecision, but there can be systematic errors. These errors can usually be handled by a detailed look at the primary data and by validation of identified mutations by Sanger sequencing. NGS can miss some mutations due to technical reasons inherent in the technology. Expansions of triplet repeats such as the ones causing myotonic dystrophy, for example, are not identifiable using NGS due to the repetitive nature of the sequence and difficulties in aligning short reads to a repetitive sequence. Further, chromosomal rearrangements and large deletions can be difficult to identify using NGS. Interpretation of variants of unknown significance (VUSs) is a problem for any genetic sequencing technology but is a particular problem when using NGS due to the sheer magnitude of the sequencing. In an exome sequencing study, an individual may have as many as 20,000 to 30,000 variants from the reference sequence. Filtering rare but benign variants from potentially pathogenic variants is difficult. A number of bioinformatic analyses are available to assess pathogenicity of variants. Locus-specific databases such as the Leiden Muscular Dystrophy pages highlight variants with known pathogenicity (www.dmd.nl). Tools such as the Single Nucleotide Polymorphism database (dbSNP), the Exome Variant Server, and 1000 Genomes Project can help to identify rare variants that have been previously identified in other populations (122–124). Analytic tools including SIFT (sorting intolerant from tolerant) and PolyPhen2 (polymorphism phenotyping) use conservation of the locus and biochemical disruption of the altered amino acid to the peptide to predict pathogenicity of a particular variant (125,126). Inclusion of DNA from parents and other family members can help to characterize the variant as benign, VUS, or pathogenic, based on segregation of the variant with the phenotype. Most commercial NGS tests include this type of segregation analysis. Taken together, these tools can be used to filter and classify variants identified in an individual or family and narrow the search for a potentially pathogenic mutation. Given the complexity of the analysis, there are a number of cases where whole genome or exome sequencing has failed to identify a mutation when it was indeed present. In these cases, the bioinformatic tools failed to properly classify a variant as pathogenic. In one recently published case, a family with clinical symptoms typical of BM presented after obtaining their own whole genome sequencing (127). Several variants were identified in genes associated with neuromuscular disorders, but none fully explained the phenotype. Reassessment of the clinical phenotype including imaging gave additional weight to the possibility of a collagen VI mutation. Traditional Sanger sequencing was performed and a pathogenic mutation in COL6A3, typical of BM was identified. In this case, the whole genome data was reanalyzed, given the known mutation in COL6A3, and it was determined that the variant was indeed present in the whole genome sequence but was misclassified by bioinformatics analysis. The authors concluded that the sequence data must be matched with careful phenotypic analysis and understanding of clinical aspects of the suspected disorder in order to come to an appropriate interpretation of the data. Current approaches to the diagnosis of neuromuscular disorders focus on careful clinical examination and application of ancillary tests to define probable disorders that are then confirmed by genetic testing. The role of clinical neurophysiology is limited primarily to distinguishing muscle disorders from nerve disorders. In the process, certain abnormalities, such as the presence of myotonic discharges can support a limited range of potential diagnoses. In recent years, the number of genetic tests has markedly increased. However, there remain disorders not diagnosed by commonly available tests. The rapid evolution of sequencing technology that allows sequencing of the entire genome or exome at a relatively low cost is likely to change diagnostic strategies in these situations. Rapid discovery of new genes involved with neuromuscular disease and broad overlap of phenotypes will prompt earlier application of broad sequencing strategies such as exome sequencing or sequencing of large targeted gene panels. While the importance of ancillary tests such as neurophysiology, muscle biopsy, and imaging will likely be diminished in diagnostic application, they will likely become increasingly important in treatment and monitoring applications. Recent advances in EIM and imaging for muscle disorders have focused on quantitative measures, which will have great application in the monitoring of treatments and progression of these disorders. 1. Arnold WD, Flanigan KM. A practical approach to molecular diagnostic testing in neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23:589–608. 2. Vasli N, Laporte J. Impacts of massively parallel sequencing for genetic diagnosis of neuromuscular disorders. Acta Neuropathol. 2013;125:173–185. 3. Fardeau M, Desguerre I. Diagnostic workup for neuromuscular diseases. Handb Clin Neurol. 2013;113:1291–1297. 4. Bonnemann CG, Wang CH, Quijano-Roy S, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014;24:289–311. 5. North KN, Wang CH, Clarke N, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord. 2014;24:97–116. 6. Norwood FL, Harling C, Chinnery PF, et al. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132:3175–3186. 7. Okada M, Kawahara G, Noguchi S, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology. 2007;69:1035–1042. 8. Clement EM, Feng L, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: Analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord. 2012;22:522–527. 9. Maggi L, Scoto M, Cirak S, et al. Congenital myopathies—clinical features and frequency of individual subtypes diagnosed over a 5-year period in the United Kingdom. Neuromuscul Disord. 2013;23:195–205. 10. Richer LP, Shevell MI, Miller SP. Diagnostic profile of neonatal hypotonia: an 11-year study. Pediatr Neurol. 2001;25:32–37. 11. Vasta I, Kinali M, Messina S, et al. Can clinical signs identify newborns with neuromuscular disorders? J Pediatr. 2005;146:73–79. 12. Carmignac V, Salih MA, Quijano-Roy S, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. 2007;61:340–351. 13. Overeem S, Schelhaas HJ, Blijham PJ, et al. Symptomatic distal myopathy with cardiomyopathy due to a MYH7 mutation. Neuromuscul Disord. 2007;17:490–493. 14. Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71:304–313. 15. Ciafaloni E, Fox DJ, Pandya S, et al. Delayed diagnosis in duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. 2009;155:380–385. 16. Crisp DE, Ziter FA, Bray PF. Diagnostic delay in Duchenne’s muscular dystrophy. JAMA. 1982;247:478–480. 17. Wong ET, Cobb C, Umehara MK, et al. Heterogeneity of serum creatine kinase activity among racial and gender groups of the population. Am J Clin Pathol. 1983;79:582–586. 18. Silvestri NJ, Wolfe GI. Asymptomatic/pauci-symptomatic creatine kinase elevations (hyperckemia). Muscle Nerve. 2013;47:805–815. 19. Kyriakides T, Angelini C, Schaefer J, et al. EFNS guidelines on the diagnostic approach to pauci- or asymptomatic hyperCKemia. Eur J Neurol. 2010;17:767–773. 21. Oh SJ. Pediatric Nerve Conduction Studies. In: Oh SJ, eds. Clinical Electromyography: Nerve Conduction Studies. Phildalphia, PA: Lippincott Williams & Wilkins; 2003: 107–135. 22. Pitt M. Paediatric electromyography in the modern world: a personal view. Dev Med Child Neurol. 2011;53:120–124. 23. Pitt M. Update in electromyography. Curr Opin Pediatr. 2013;25:676–681. 24. Bady B, Vila A, Boulliat G, et al. [Value of electromyography in the child. Apropos of 1,624 examinations performed over a 3 year period]. Rev Electroencephalogr Neurophysiol Clin. 1983;13:282–288. 25. Hellmann M, von Kleist-Retzow JC, Haupt WF, et al. Diagnostic value of electromyography in children and adolescents. J Clin Neurophysiol. 2005;22:43–48. 26. Rabie M, Jossiphov J, Nevo Y. Electromyography (EMG) accuracy compared to muscle biopsy in childhood. J Child Neurol. 2007;22:803–808. 27. Paganoni S, Amato A. Electrodiagnostic evaluation of myopathies. Phys Med Rehabil Clin N Am. 2013;24:193–207. 28. Ghosh PS, Sorenson EJ. Diagnostic Yield of Electromyography in Children With Myopathic Disorders. Pediatr Neurol. 2014; 51(2):215–219. 29. Shah DU, Darras BT, Markowitz JA, et al. The spectrum of myotonic and myopathic disorders in a pediatric electromyography laboratory over 12 years. Pediatr Neurol. 2012;47:97–100. 30. Pitt MC. Nerve conduction studies and needle EMG in very small children. Eur J Paediatr Neurol. 2012;16:285–291. 31. Darras BT, Jones HR. Diagnosis of pediatric neuromuscular disorders in the era of DNA analysis. Pediatr Neurol. 2000;23:289–300. 32. Shorer Z, Philpot J, Muntoni F, et al. Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J Child Neurol. 1995;10:472–475. 33. Mercuri E, Pennock J, Goodwin F, et al. Sequential study of central and peripheral nervous system involvement in an infant with merosin-deficient congenital muscular dystrophy. Neuromuscul Disord. 1996;6:425–429. 34. Quijano-Roy S, Renault F, Romero N, et al. EMG and nerve conduction studies in children with congenital muscular dystrophy. Muscle Nerve. 2004;29:292–299. 35. Fujii Y, Sugiura C, Fukuda C, et al. Sequential neuroradiological and neurophysiological studies in a Japanese girl with merosin-deficient congenital muscular dystrophy. Brain Dev. 2011;33:140–144. 36. Peric S, Stojanovic VR, Nikolic A, et al. Peripheral neuropathy in patients with myotonic dystrophy type 1. Neurol Res. 2013;35:331–335. 37. Zanette G, Manani G, Pittoni G, et al. Prevalence of unsuspected myopathy in infants presenting for clubfoot surgery. Paediatr Anaesth. 1995;5:165–170. 38. Rutkove SB. Electrical impedance myography: Background, current state, and future directions. Muscle Nerve. 2009;40:936–946. 39. Rutkove SB, Shefner JM, Gregas M, et al. Characterizing spinal muscular atrophy with electrical impedance myography. Muscle Nerve. 2010;42:915–921. 40. Rutkove SB, Darras BT. Electrical impedance myography for the assessment of children with muscular dystrophy: a preliminary study. J Phys Conf Ser. 2013;434. 41. Dubowitz V, Sewry CA. Muscle biopsy: a practical approach. 3rd ed. Philadelphia, PA: Saunders/Elsevier; 2007. 42. Heckmatt JZ, Dubowitz V. Ultrasound imaging and directed needle biopsy in the diagnosis of selective involvement in muscle disease. J Child Neurol. 1987;2:205–213. 43. Joyce NC, Oskarsson B, Jin LW. Muscle biopsy evaluation in neuromuscular disorders. Phys Med Rehabil Clin N Am. 2012;23:609–631. 44. Tarnopolsky MA, Pearce E, Smith K, Lach B. Suction-modified Bergstrom muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve. 2011;43:717–725. 45. Jaradeh SS, Ho H. Muscle, nerve, and skin biopsy. Neurol Clin. 2004;22:539–561 46. Meola G, Bugiardini E, Cardani R. Muscle biopsy. J Neurol. 2012;259:601–610. 48. Wallefeld W, Krause S, Nowak KJ, et al. Severe nemaline myopathy caused by mutations of the stop codon of the skeletal muscle alpha actin gene (ACTA1). Neuromuscul Disord. 2006;16:541–547. 49. Heckmatt JZ, Dubowitz V, Leeman S. Detection of pathological change in dystrophic muscle with B-scan ultrasound imaging. Lancet. 1980;1:1389–1390. 50. Heckmatt JZ, Leeman S, Dubowitz V. Ultrasound imaging in the diagnosis of muscle disease. J Pediatr. 1982;101:656–660. 51. Pillen S, Arts IM, Zwarts MJ. Muscle ultrasound in neuromuscular disorders. Muscle Nerve. 2008;37:679–693. 52. Bonnemann CG, Brockmann K, Hanefeld F. Muscle ultrasound in Bethlem myopathy. Neuropediatrics. 2003;34:335–336. 53. Bharucha-Goebel DX, Santi M, Medne L, et al. Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology. 2013;80:1584–1589. 54. Pillen S, van Alfen N. Skeletal muscle ultrasound. Neurol Res. 2011;33:1016–1024. 55. Wattjes MP, Kley RA, Fischer D. Neuromuscular imaging in inherited muscle diseases. Eur Radiol. 2010;20:2447–2460. 56. Mercuri E, Talim B, Moghadaszadeh B, et al. Clinical and imaging findings in six cases of congenital muscular dystrophy with rigid spine syndrome linked to chromosome 1p (RSMD1). Neuromuscul Disord. 2002;12:631–638. 57. Kornblum C, Lutterbey G, Bogdanow M, et al. Distinct neuromuscular phenotypes in myotonic dystrophy types 1 and 2: a whole body highfield MRI study. J Neurol. 2006;253:753–761. 58. Fischer D, Kley RA, Strach K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology. 2008;71:758–765. 59. Fischmann A, Hafner P, Gloor M, et al. Quantitative MRI and loss of free ambulation in Duchenne muscular dystrophy. J Neurol. 2013;260:969–974. 60. Willis TA, Hollingsworth KG, Coombs A, et al. Quantitative muscle MRI as an assessment tool for monitoring disease progression in LGMD2I: a multicentre longitudinal study. PLoS One. 2013;8:e70993. 61. Arpan I, Willcocks RJ, Forbes SC, et al. Examination of effects of corticosteroids on skeletal muscles of boys with DMD using MRI and MRS. Neurology. 2014;83:974–980. 62. Costa AF, Di Primio GA, Schweitzer ME. Magnetic resonance imaging of muscle disease: a pattern-based approach. Muscle Nerve. 2012;46:465–481. 63. Quijano-Roy S, Carlier RY, Fischer D. Muscle imaging in congenital myopathies. Semin Pediatr Neurol. 2011;18:221–229. 64. Mercuri E, Clements E, Offiah A, et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol. 2010;67:201–208. 65. ten Dam L, van der Kooi AJ, van Wattingen M, et al. Reliability and accuracy of skeletal muscle imaging in limb-girdle muscular dystrophies. Neurology. 2012;79:1716–1723. 66. Quijano-Roy S, Avila-Smirnow D, Carlier RY. Whole body muscle MRI protocol: pattern recognition in early onset NM disorders. Neuromuscul Disord. 2012;22 Suppl 2:S68–S84. 67. Tasca G, Monforte M, Iannaccone E, et al. Upper girdle imaging in facioscapulohumeral muscular dystrophy. PLoS One. 2014;9:e100292. 68. Hollingsworth KG, Garrood P, Eagle M, Bushby K, Straub V. Magnetic resonance imaging in Duchenne muscular dystrophy: longitudinal assessment of natural history over 18 months. Muscle Nerve. 2013;48:586–588. 69. Kinali M, Arechavala-Gomeza V, Cirak S, et al. Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology. 2011;76:346–353. 70. Tasca G, Iannaccone E, Monforte M, et al. Muscle MRI in Becker muscular dystrophy. Neuromuscul Disord. 2012;22 Suppl 2:S100–S106. 71. Jungbluth H, Davis MR, Muller C, et al. Magnetic resonance imaging of muscle in congenital myopathies associated with RYR1 mutations. Neuromuscul Disord. 2004;14:785–790. 72. Fischer D, Herasse M, Ferreiro A, et al. Muscle imaging in dominant core myopathies linked or unlinked to the ryanodine receptor 1 gene. Neurology. 2006;67:2217–2220. 74. Mercuri E, Lampe A, Allsop J, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15:303–310. 75. Lovitt S, Moore SL, Marden FA. The use of MRI in the evaluation of myopathy. Clin Neurophysiol. 2006;117:486–495. 76. Mercuri E, Bushby K, Ricci E, et al. Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contractures. Neuromuscul Disord. 2005;15:164–171. 77. Fischer D, Walter MC, Kesper K, et al. Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. J Neurol. 2005;252:538–547. 78. Koenig M, Hoffman EP, Bertelson CJ, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. 79. Kaplan JC, Hamroun D. The 2014 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord. 2013;23:1081–1111. 80. Wang CH, Dowling JJ, North K, et al. Consensus statement on standard of care for congenital myopathies. J Child Neurol. 2012;27:363–382. 81. Moxley RT, 3rd, Pandya S, Ciafaloni E, et al. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J Child Neurol. 2010;25:1116–1129. 82. Gagnon C, Chouinard MC, Laberge L, et al. Health supervision and anticipatory guidance in adult myotonic dystrophy type 1. Neuromuscul Disord. 2010;20:847–851. 83. Wang CH, Finkel RS, Bertini ES, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22:1027–1049. 84. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. 85. Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010;81:358–367. 86. Bonnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol. 2011;7:379–390. 87. Toda T, Kobayashi K, Kondo-Iida E, Sasaki J, Nakamura Y. The Fukuyama congenital muscular dystrophy story. Neuromuscul Disord. 2000;10:153–159. 88. Watanabe M, Kobayashi K, Jin F, et al. Founder SVA retrotransposal insertion in Fukuyama-type congenital muscular dystrophy and its origin in Japanese and Northeast Asian populations. Am J Med Genet A. 2005;138:344–348. 89. . Davis MR, Haan E, Jungbluth H, et al. Principal mutation hotspot for central core disease and related myopathies in the C-terminal transmembrane region of the RYR1 gene. Neuromuscul Disord. 2003;13:151–157. 90. Flanigan KM, von Niederhausern A, Dunn DM, et al. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72:931–939. 91. Klein A, Lillis S, Munteanu I, et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat. 2012;33:981–988. 92. Chauveau C, Rowell J, Ferreiro A. A Rising Titan: TTN Review and Mutation Update. Hum Mutat. 2014; 35(9):1046–1059. 93. Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492–500. 94. Jimenez-Mallebrera C, Maioli MA, Kim J, et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul Disord. 2006;16:571–582. 96. Bethlem J, Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain. 1976;99:91–100. 97. Jobsis GJ, Keizers H, Vreijling JP, et al. Type VI collagen mutations in Bethlem myopathy, an auto-somal dominant myopathy with contractures. Nat Genet. 1996;14:113–115. 98. Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci U S A. 2001;98:7516–7521. 99. Butterfield RJ, Foley AR, Dastgir J, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat. 2013;34:1558–1567. 100. Bonne G, Quijano-Roy S. Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb Clin Neurol. 2013;113:1367–1376. 101. Bonne G, Leturcq F, Ben Yaou R. Emery-Dreifuss Muscular Dystrophy. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews [Intermet]. Seattle, WA: Univeristy of Washington; 2013. 102. Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30:1657–1666. 103. Dent KM, Dunn DM, von Niederhausern AC, et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet A. 2005;134:295–298. 104. Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014; 50(4):477–487. 105. Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647. 106. Lee T, Takeshima Y, Kusunoki N, et al. Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J Hum Genet. 2014;59:46–50. 107. Helderman-van den Enden AT, de Jong R, den Dunnen JT, et al. Recurrence risk due to germ line mosaicism: Duchenne and Becker muscular dystrophy. Clin Genet. 2009;75:465–472. 108. Soltanzadeh P, Friez MJ, Dunn D, et al. Clinical and genetic characterization of manifesting carriers of DMD mutations. Neuromuscul Disord. 2010;20:499–504. 109. Schade van Westrum SM, Hoogerwaard EM, Dekker L, et al. Cardiac abnormalities in a follow-up study on carriers of Duchenne and Becker muscular dystrophy. Neurology. 2011;77:62–66. 110. Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: the management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th–9th June 2002, Naarden, the Netherlands. Neuromuscul Disord. 2003;13:166–172. 111. Consortium IHGS. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. 112. Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). www.genome.gov/sequencingcosts. Accessed August 2014. 113. Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet. 2010;11:31–46. 114. Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362:1181–1191. 115. Hayden EC. Technology: The $1,000 genome. Nature. 2014;507:294–295. 116. Mardis ER. The $1,000 genome, the $100,000 analysis? Genome Med. 2010;2:84. 117. Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. 118. Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. 119. Chilamakuri CS, Lorenz S, Madoui MA, et al. Performance comparison of four exome capture systems for deep sequencing. BMC Genomics. 2014;15:449. 120. Nigro V, Piluso G. Next generation sequencing (NGS) strategies for the genetic testing of myopathies. Acta Myol. 2012;31:196–200. 121. Vasli N, Bohm J, Le Gras S, et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol. 2012;124:273–283. 123. Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. 124. Exome Variant Server. In. 2014 ed: NHLBI GO Exome Sequencing Project (ESP). Seattle, WA; 2014. 125. Ng PC, Henikoff S. Accounting for human polymorphisms predicted to affect protein function. Genome Res. 2002;12:436–446. 126. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. 127. Foley AR, Pitceathly RD, He J, et al. Whole-genome sequencing and the clinician: a tale of two cities. J Neurol Neurosurg Psychiatry. 2014;85:1012–1015.
Pediatric Muscular Dystrophies and Myopathies: The Role of Neurophysiology, Genetics, and Ancillary Testing
CLINICAL EVALUATION/EXAMINATION
ANCILLARY STUDIES
Creatine Kinase
Clinical Neurophysiology
Electrical Impedance Myography
Muscle Biopsy
Muscle Imaging
Ultrasound
Computed Tomography
Magnetic Resonance Imaging
Imaging Findings in Specific Disorders
Duchenne and Becker Muscular Dystrophies
Congenital Myopathies/Muscular Dystrophies
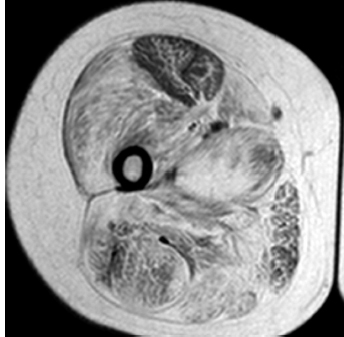
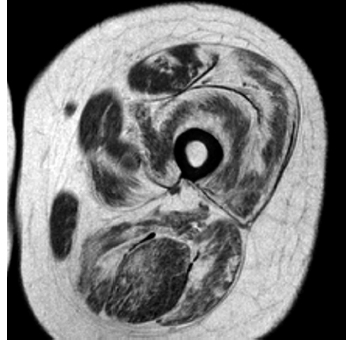
Limb-Girdle Muscular Dystrophy
GENETIC TESTING
Complications of Genetic Testing in Neuromuscular Disorders
Genetic and Phenotypic Heterogeneity
Duchenne and Becker Muscular Dystrophy
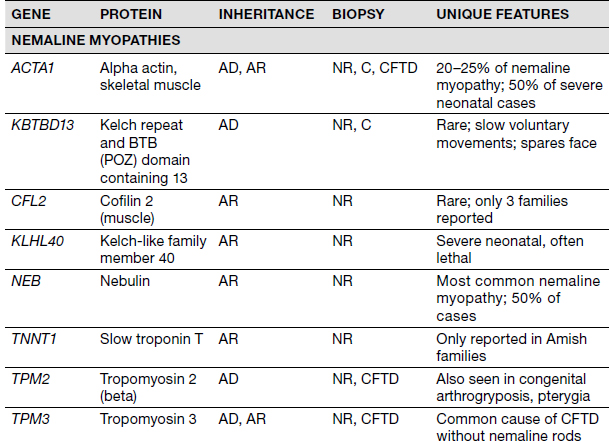
Congenital Myopathies
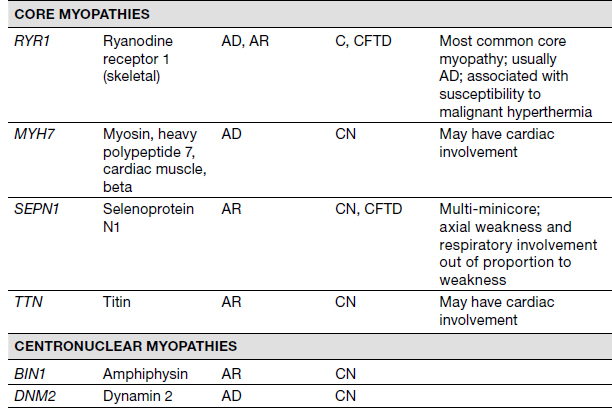

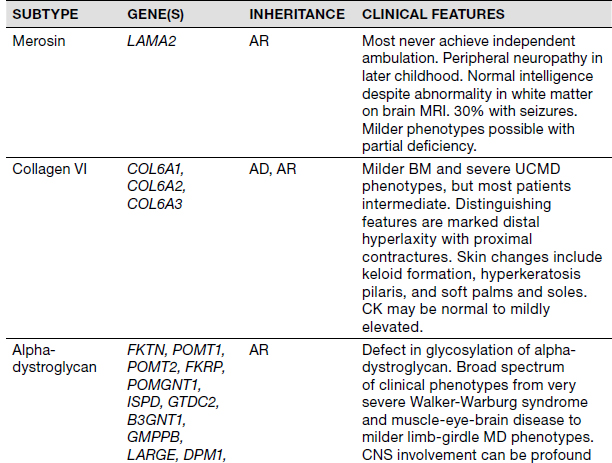
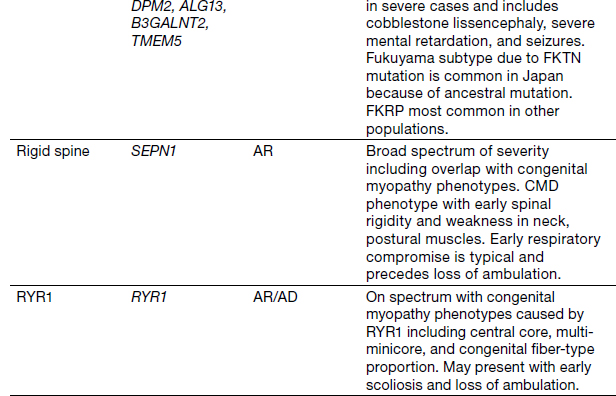
Congenital Muscular Dystrophies

Next-Generation Sequencing
Targeted Gene and Exome Sequencing
Difficulties of NGS
CONCLUSION
REFERENCES
Related posts:
 Clinical Neurophysiology in Pediatric Peripheral Neuropathy
Intraoperative Considerations in the Pediatric Patient
Clinical Neurophysiology in Pediatric Peripheral Neuropathy
Intraoperative Considerations in the Pediatric Patient
 Clinical Evaluation in Pediatric Peripheral Neuropathies
Clinical Evaluation in Pediatric Peripheral Neuropathies
 Autonomic Disorders in Children
Autonomic Disorders in Children
 EEG Interpretation in Childhood Epilepsies
EEG Interpretation in Childhood Epilepsies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


