Pediatric Pituitary Adenomas
Although relatively uncommon, pituitary adenomas account for 2 to 6% of pediatric brain tumors.1 Of all pituitary tumors, only 3.5 to 8.5% are diagnosed by the age of 20, and only 25% of those present before the age of 12 (primarily corticotropinomas).2 Adenomas that secrete adrenocorticotropic hormone (ACTH) and growth hormone (GH) are the most common type of adenomas in prepubertal children. Prolactinomas predominate in the pubertal and postpubertal populations.3–7 Prolactin-secreting tumors account for 30 to 50% of pediatric cases, and ACTH-secreting tumors also account for 30 to 50% of pediatric cases. GH-secreting tumors comprise 10% in most series and are the most common type occurring in infants.3,6,8 Although nonfunctioning adenomas account for approximately one-third of adult adenomas, they constitute only 5% of pediatric adenomas.3,9,10 Thyrotroph- and gonadotroph-secreting adenomas are extremely rare and have been reported in only a few cases. Although they are often sporadic, they can occur in the context of multiple endocrine neoplasia type 1 (MEN-1), McCune-Albright syndrome, Carney complex, or familial isolated pituitary adenomas (FIPAs).11
38.1 Indications for Treatment: Medical and Surgical
Presenting signs and symptoms are generally related to endocrine dysfunction rather than to ophthalmologic complaints but vary according to maturity of the patient. Prepubertal children and pubescent boys typically present with headaches, visual complaints, and growth delay. Pubescent girls present with pubertal arrest and hypogonadism with or without galactorrhea. Fortunately, pituitary apoplexy is a rare event in patients with pituitary adenomas, and surgical emergencies are therefore uncommon in the pediatric population.12–14 Neurosurgical emergencies include increased intracranial pressure and rapid visual deterioration that may require emergent drainage of the ventricles (in the case of hydrocephalus) or cyst contents in order to stabilize the patient.
The evaluation of a pituitary mass includes contrast-enhanced magnetic resonance (MR) imaging with dedicated pituitary imaging, a full biochemical evaluation of the hypothalamic–pituitary axis, and an ophthalmologic evaluation (see box “▶ Preoperative Evaluation of a Pituitary Adenoma”). Computed tomography (CT) may also be helpful to assess the bony architecture and degree of pneumatization of the sphenoid sinus. CT may also help in the differential diagnosis of pituitary adenoma versus a craniopharyngioma. Stress-dose corticosteroids should be considered for any patient with a large pituitary mass who is experiencing physiologic stress.
Preoperative Evaluation of a Pituitary Adenoma
Imaging
Magnetic resonance imaging + gadolinium with pituitary sequences
Computed tomography for bony architecture
Vision
Formal neuro-ophthalmologic examination
Visual fields
Endocrine
Adrenocorticotropic hormone (ACTH)
Cortisol (a.m.)
Follicle-stimulating hormone (FSH)
Luteinizing hormone (LH)
Growth hormone (GH), insulin-like growth factor-1 (IGF-1)
Prolactin
Thyroid-stimulating hormone (TSH), free thyroxine (T4)
Basic metabolic panel
Urine specific gravity and sodium
Source: Adapted from Kiehna EN, Payne SC, Jane JA Jr. Surgical treatment of Rathke’s cleft cysts. In: Laws ER Jr, Sheehan JP, eds. Sellar and Parasellar Tumors. New York, NY: Thieme Medical Publishers; 2011:chap 10.
38.1.1 Prolactinomas
Prolactinomas are the most common pituitary tumor, comprising nearly 50% of pituitary tumors in children. They generally present at the time of or after puberty with primary or secondary amenorrhea in girls.15–17 Males more frequently have macroprolactinomas, which are associated with a higher incidence of neurologic and ophthalmologic signs.11 The diagnosis is confirmed by measuring the serum prolactin, with values greater than 200 ng/mL diagnostic of a prolactinoma.
Medical therapy is the mainstay of treatment for these tumors. Prolactinomas should first be treated with dopamine agonists (e.g., bromocriptine, cabergoline), which are effective in normalizing the PRL levels and shrinking the tumor mass in the majority of children and adolescents (▶ Fig. 38.1).3,16,18 Cabergoline is often favored over bromocriptine because it has a longer half-life and can be administered on a weekly basis to normalize the serum prolactin levels and restore gonadal function. It has also proved to be more effective in patients whose tumors are poorly responsive or resistant to other agents.18–24 If patients cannot tolerate the medications or if their tumors are refractory to the medications, surgical remission may be obtained in 85% of patients with microprolactinomas. Lower rates of remission are expected in those with macroadenomas and those with tumors invading the cavernous sinus. However, surgical debulking may reduce the tumor burden enough to allow either radiosurgery or the recapture of effective medical therapy.
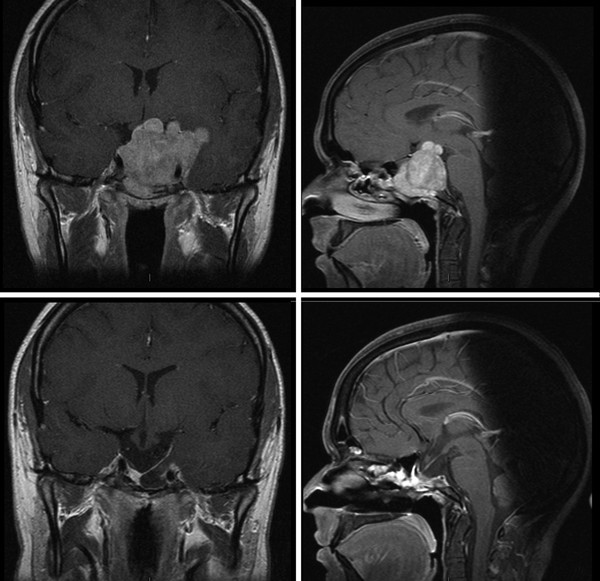
Fig. 38.1 Prolactinoma before (above) and 3 weeks after (below) medical treatment with cabergoline. Vision returned to normal within 1 week of the initiation of medical therapy.
38.1.2 Corticotropinomas
Corticotropinomas (Cushing disease) typically appear in prepubescent children between 11 and 15 years of age and are a frequent cause of adrenal hyperfunction in this age group.25–27 Weight gain is the common hallmark; this is accompanied by growth failure; premature puberty; facial plethora; atrophic striae in the abdomen, legs, and arms; muscular weakness; hypertension; and osteoporosis. Children may also have impaired carbohydrate tolerance (although frank diabetes mellitus is uncommon). Increases in adrenal androgens may cause acne and excessive hair growth. Corticotropinomas are diagnosed through the measurement of basal and stimulated levels of cortisol and ACTH (▶ Fig. 38.2).
Cushing syndrome must first be confirmed on the basis of serially elevated 24-hour urinary free cortisol (corrected for the child’s body surface area) or 11 p.m. salivary cortisol measurements. Administration of low dose of dexamethasone at midnight (15 µg/kg) does not induce suppression of morning serum cortisol concentrations in patients with Cushing syndrome. Cushing disease must then be distinguished from ectopic ACTH-producing lesions. Suppression of cortisol by more than 50% after the administration of high-dose dexamethasone at midnight (120 µg/kg) will confirm that hypercortisolism is due to an ACTH-secreting pituitary adenoma.28 In many cases in which neuroimaging is not diagnostic, inferior petrosal sinus sampling can have a high specificity both for diagnosing a pituitary location with ACTH production (sensitivity of 97%) and defining laterality (in 75% of cases), but it carries a high rate of false-positive results.
Cushing disease is preferentially treated by transsphenoidal resection; this results in a surgical remission in the majority of children, with initial remission rates of 70 to 98% and long-term remission rates of 50 to 98% with multimodal therapy.1,3,8,25,26,29–33
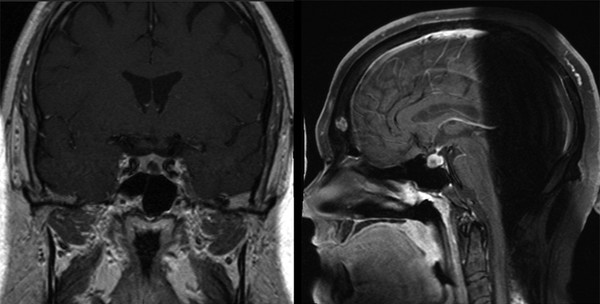
Fig. 38.2 Corticotropinoma. Coronal and sagittal T1-weighted gadolinium-enhanced images revealing a left-sided intrasellar adenoma.
38.1.3 Somatotropinomas
Somatotropinomas (acromegaly) are rare in children (5 to 15% of pituitary tumors) but are notable for causing gigantism before growth plate fusion, diabetes, visual disturbances, and/or headaches.3 These tumors may occasionally concomitantly secrete prolactin and thyroid hormone, resulting in additional symptomatology. They may be associated with McCune-Albright syndrome or Carney complex as a result of somatotroph hyperplasia.11 Somatotropinomas are usually diagnosed clinically but are confirmed by measuring circulating concentrations of insulin-like growth factor-1 (IGF-1,) which correlate with the integrated 24-hour GH secretion levels (▶ Fig. 38.3). Further evaluation includes an oral glucose tolerance test; failure of GH suppression or a paradoxical increase in GH production identifies patients with a GH-secreting pituitary lesion.34
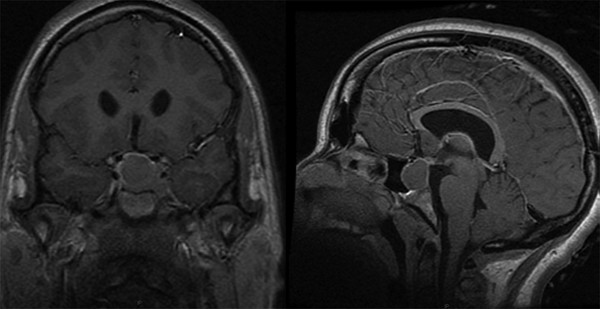
Fig. 38.3 Somatotropinoma. Coronal and sagittal T1-weighted gadolinium-enhanced images revealing a right-sided intrasellar adenoma. Note the frontal bossing and expanded diploic space on the sagittal image.
Acromegaly has traditionally been treated primarily with transsphenoidal surgery. In most surgical series, approximately 50 to 70% of patients with acromegaly achieve normal IGF-1 levels.35–37 Treatment with somatostatin analogues (e.g., octreotide, lanreotide) has been applied to the adult population, and the growth hormone receptor antagonist pegvisomant has been effectively used alone or in combination with somatostatin analogues. There is little if any experience in children with these agents.
38.1.4 Thyrotropinomas
Thyrotropinomas are extremely rare but usually present as macroadenomas with mass effect symptoms, such as headache and visual disturbance, together with various symptoms and signs of hyperthyroidism.38,39 They are confirmed through thyroid function tests. A failure to respond to a thyroid-releasing hormone (TRH) stimulation test distinguishes a thyrotropinoma from central thyroxine (T4) resistance. An elevated α-subunit level relative to the thyroid-stimulating hormone (TSH) level may also be useful.38,39
Thyotropinomas are primarily treated with transsphenoidal resection; however, they often require adjuvant radiation therapy. These tumors may also respond to somatostatin analogues.
38.1.5 Nonsecreting Adenomas
Nonsecreting adenomas are uncommon in childhood because they are slow-growing and often take decades before signs of pituitary insufficiency and mass effect are evident.40 Apoplexy is a rare event in the pediatric population and is most common with nonfunctioning macroadenomas (up to 21%). Cystic formation aside from hemorrhage may occur in up to 17%.40
38.2 Surgical Treatment
There are certain challenges to transsphenoidal surgery in children, including small nares, narrow passageways, and poor pneumatization of the sphenoid sinus. The sphenoid sinus begins to become pneumatized at about 3 years of age, and pneumatization proceeds rapidly until age 7; the sinus reaches its average normal capacity of 7.5 mL in adolescence. In approximately 5% of patients, the sphenoid bone remains minimally pneumatized (▶ Fig. 38.4). Because of this lack of pneumatization and the shallowness of a child’s sella turcica relative to that of an adult, transsphenoidal approaches in children often require substantial drilling of the sphenoid bone, particularly in the case of microadenomas.
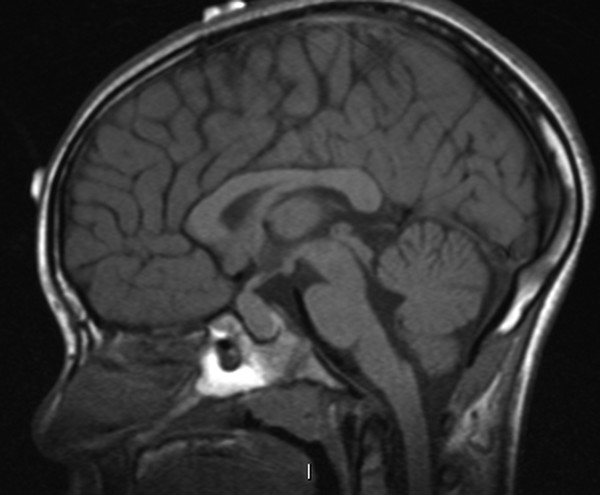
Fig. 38.4 Incomplete pneumatization of the sphenoid sinus in an 8-year-old with a pituitary mass.









