Phenobarbital and Other Barbiturates
Mervyn J. Eadie
Patrick Kwan
Introduction
The antiepileptic effect of phenobarbital (phenobarbitone) was described by Alfred Hauptmann as long ago as 1912,29 a little more than half a century after Locock, in 1857, had recognized what proved to be the first effective antiepileptic agent, potassium bromide. Phenobarbital has been in continuous use as an antiepileptic agent for nearly a century. In recent years, its popularity in developed countries has declined, although it and its congener primidone still account for some 14% of all antiepileptic drug (AED) use in Britain.52 It is more widely used in less affluent societies and, on a global scale, remains one of the most important AEDs. Phenobarbital possesses a broad spectrum of antiepileptic activity and an effectiveness similar to that of many other marketed AEDs. It is safe, cheap, and available in convenient oral and parenteral dosage forms. These advantages have led some investigators to call for renewed scientific study to enhance its proper use in the modern therapeutic era.42
Attempts have been made to modify the phenobarbital molecule to derive agents with greater efficacy and lesser toxicity. Primidone (desoxyphenobarbital) has gained a place as an established antiepileptic agent in its own right (see Chapter 158). However, at least part of its pharmacologic action is effected via phenobarbital, to which it is metabolized in the body. The N-methyl analog of phenobarbital, mephobarbital (N-methylphenobarbitone), was introduced into therapeutics by Blum in 1932.8 In some places, it was preferred to phenobarbital although its use appears to be declining. Phenobarbital and mephobarbital analogs that lack an aromatic ring on the 5-carbon atom of the barbiturate ring (barbital and metharbital) also possess antiepileptic properties. However, few data concerning them are available, and they now appear to be little used. Eterobarb (1,3-dimethoxymethylphenobarbital) underwent some modern scientific studies, but thereafter it did not seem to achieve any widespread clinical use. Only phenobarbital, and to an extent mephobarbital, will be discussed further.
Chemistry, Formulations, Assay
Chemistry
Phenobarbital (5-ethyl-5-phenylbarbituric acid) is a white crystalline material with a pKa of 7.2. Mephobarbital (5-ethyl-1-methyl-5-phenylbarbituric acid) is chiral at the carbon atom at the 5-position of the barbiturate ring by virtue of the methyl group substituent on the 1-position of the barbituric acid molecule. It is supplied commercially as a racemate. The racemic material is more lipophilic than phenobarbital and has a pKa value of 7.8.
Figure 1 shows the formulas and metabolic pathways for phenobarbital (5-ethyl-5-phenylbarbituric acid), r-(–)-mephobarbital, and s-(+)-mephobarbital.
Formulations
Phenobarbital has been marketed either as the acid or the sodium salt in 15-, 30-, 50-, 60-, and 100-mg tablets. However, not all strengths may be available in all countries. Oral solutions of various strengths containing sodium phenobarbital can be prepared. Parenteral solutions of the drug usually contain 100 or 200 mg of the sodium salt.
Mephobarbital has been marketed as 30-, 32-, 50-, 60-, 100-, and 200-mg tablets.
Assay
Phenobarbital in biologic fluids has been measured by ultraviolet spectrophotometry, gas and liquid chromatography, and various immunoassay methods. Mephobarbital is usually measured by gas or liquid chromatography, although methyl derivative formation for gas chromatography results in any phenobarbital that is present after biotransformation being measured as mephobarbital.
Pharmacology
Mephobarbital is biotransformed to phenobarbital, and it is believed that most of the anticonvulsant action of mephobarbital depends on this metabolic product. However, there is some indirect evidence that intact R,S-mephobarbital probably has an antiepileptic effect14 whose mechanism is not established. Only the mechanism of action of phenobarbital is considered further in this chapter.
Seizure Models
Inconsistent reports are available of phenobarbital’s effects on epileptic activity in experimental seizure foci in animals.23 The drug appears to limit the spread of activity from such foci, probably by interfering with synaptic neurotransmission.25 Consistent with its clinical effect against generalized tonic–clonic seizures, phenobarbital is effective in the maximal electroshock model. It is also active against pentylenetetrazol-induced seizures, although it has no effect against human absence seizures.81
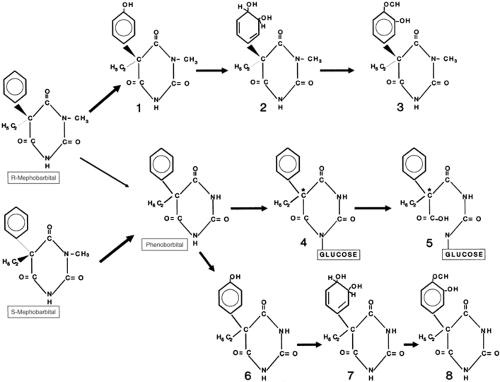 FIGURE 1. Chemical formulas and known metabolic pathways for mephobarbital and phenobarbital. 1, p-Hydroxy derivative of R-mephobarbital. 2, Diol derivative of R-mephobarbital. 3, O-Methyl catechol derivative of R-mephobarbital. 4, Phenobarbitone-N-glucoside (the asymmetric carbon atom is marked with an asterisk, but the structures of the separate enantiomers are not drawn). 5, Barbiturate ring-opened derivatives of phenobarbitone-N-glucoside. 6, p-Hydroxy derivative of phenobarbital. 7, Diol derivative of phenobarbital. 8, O-Methyl catechol derivative of phenobarbital. Many of the metabolites containing a hydroxyl group are excreted in part as glucuronides. |
Mechanism of Action
Although a number phenobarbital’s biochemical actions have been described, such as inhibition of mitochondrial electron transport, some are significant only at drug concentrations
appreciably higher than those at which an antiepileptic effect occurs in clinical use. It is thought that phenobarbital acts as an anticonvulsant mainly by prolonging the opening of the postsynaptic cell membrane chloride (Cl–) channel, a component of the γ-aminobutyric acid (GABA)-A receptor.5 The resultant increase in Cl– flux tends to hyperpolarize the postsynaptic neuronal cell membrane, thus impeding the transmission of epileptic activity. At concentrations higher than the usual human therapeutic ones, it may also affect voltage-dependent sodium (Na+) channels to diminish rapid neuronal firing.47
appreciably higher than those at which an antiepileptic effect occurs in clinical use. It is thought that phenobarbital acts as an anticonvulsant mainly by prolonging the opening of the postsynaptic cell membrane chloride (Cl–) channel, a component of the γ-aminobutyric acid (GABA)-A receptor.5 The resultant increase in Cl– flux tends to hyperpolarize the postsynaptic neuronal cell membrane, thus impeding the transmission of epileptic activity. At concentrations higher than the usual human therapeutic ones, it may also affect voltage-dependent sodium (Na+) channels to diminish rapid neuronal firing.47
Clinical Pharmacology
Absorption
Phenobarbital is virtually fully bioavailable after oral administration54 and appears to be absorbed fairly rapidly. Eadie et al.21 calculated the mean absorption half-time as 1.37 hours. It has sometimes been stated that the drug is slowly absorbed, but this assertion appears to be based on the finding that the peak plasma concentration did not occur until some 6 to 18 hours after intake.46 However, the time of maximal concentration (Tmax) of a drug occurs when its elimination rate equals its absorption rate. The slow elimination of phenobarbital could have accounted for its late Tmax. More recent studies suggest a Tmax of 0.5 to 4 hours after oral administration, compared with 2 to 8 hours after intramuscular injection.54,80 Generally little difference is noted in Tmax or bioavailability between the oral and intramuscular routes of administration.78
The oral dose of mephobarbital on a molar basis must be approximately twice that of phenobarbital to achieve the same steady-state plasma concentration of the latter. Because of this, it has sometimes been assumed that mephobarbital has only a 50% oral bioavailability. However, Hooper et al.33 showed in two subjects that oral R,S-mephobarbital was 75% bioavailable, and in further work showed that the oral clearance of R-mephobarbital (approximately 0.5 L/kg per hour) was high enough to suggest that this enantiomer of the drug probably underwent significant presystemic elimination,34 at least in extensive metabolizers of the enantiomer. Therefore, the reduced oral bioavailability of R,S-mephobarbital is probably due to appreciable presystemic metabolism rather than incomplete
absorption. The absorption half-time for R,S-mephobarbital was 0.48 and 0.38 hours in two subjects,33 and the mean Tmax for R-mephobarbital was 2.29 ± SD 1.03 hours and that for S-mephobarbital was 3.50 ± SD 1.52 hours.21 However, the absorption parameters of R,S-mephobarbital and its individual enantiomers are less relevant clinically than the time (around 4 or 5 days) when peak plasma concentrations of derived phenobarbital occur after a dose of mephobarbital.
absorption. The absorption half-time for R,S-mephobarbital was 0.48 and 0.38 hours in two subjects,33 and the mean Tmax for R-mephobarbital was 2.29 ± SD 1.03 hours and that for S-mephobarbital was 3.50 ± SD 1.52 hours.21 However, the absorption parameters of R,S-mephobarbital and its individual enantiomers are less relevant clinically than the time (around 4 or 5 days) when peak plasma concentrations of derived phenobarbital occur after a dose of mephobarbital.
Distribution
The value of the apparent volume of distribution of phenobarbital in humans (approximately 0.5 to 0.7 L/kg) suggests that the drug is distributed throughout body water, with little selective regional or tissue concentration. Some 50% to 60% of the phenobarbital in plasma is protein-bound. Concentrations of phenobarbital in the cerebrospinal fluid correlate with its unbound serum levels.46 In vivo studies in rats have shown that phenobarbital is a substrate of the efflux drug transporter P-glycoprotein, which is expressed at cerebral capillary endothelium. The clinical relevance of this remains to be determined, but growing evidence suggests that, by extruding AEDs from their intended site of action in the brain, overexpression of P-glycoprotein could potentially play an important role in the pathogenesis of pharmacoresistance in epilepsy.43
Compared with phenobarbital, R,S-mephobarbital has a higher apparent volume of distribution (mean, 132 L in adults).20 This value correlates with the greater lipophilicity of the drug, suggesting that it may achieve higher simultaneous concentrations in tissues than in plasma. Craig and Shideman14 found brain concentrations of R,S-mephobarbital were about eight times its simultaneous blood concentrations in rats. The mean apparent volume of distribution of R– and S-mephobarbital was 5.32 ± SD 3.33 L/kg and 1.73 ± SD 0.31 L/kg, respectively,44 but the value for the former may have been overestimated. Approximately 67% of R-mephobarbital and 59% of S-mephobarbital in plasma are protein bound.44
Metabolism
Figure 1 depicts a metabolic schema for both barbiturate derivatives.
Phenobarbital
At steady state, a mean of 25% to 27% of a phenobarbital dose is excreted unmetabolized in urine.7,82 Because the pKa of phenobarbital (7.2) is slightly above the usual pH of urine, any increase in the alkalinity of urine is likely to increase the ionized fraction of the drug in urine and the amount of intact phenobarbital excreted.
The majority of a phenobarbital dose is eliminated by hepatic metabolism via hydroxylation and glucosidation, with wide interindividual variability in the contributions from these routes. Hydroxylation is attributed to the cytochrome P450 (CYP) enzyme system, yielding a phenolic derivative (p-hydroxyphenobarbital), most of which is then conjugated, mainly with glucuronic acid, before being excreted in urine. This phenolic metabolite is thought not to be biologically active. It accounted for 16 ± SD 10% of the drug dose in the steady-state metabolic balance study of Bernus et al.7 There is uncertainty over the precise CYP isoenzymes involved, but they would appear to include CYP2C9, with minor contributions from CYP2C19 and CYP2E1.1 Reports on the influence of genetic polymorphism of CYP2C19 on the clearance of phenobarbital are conflicting.28,49 Whether variation of the CYP2C9 gene affects the overall metabolism of phenobarbital has not been reported.
The urinary excretion of unmetabolized phenobarbital and its phenolic metabolite (conjugated and nonconjugated) accounts for approximately 40% of a drug dose.7,82 In addition, phenobarbital undergoes N-glucosidation,56,71 yielding R– and S-enantiomers of the glucoside. Bernus et al.7 found that, under steady-state conditions, 14 ± SD 11% and up to 4% of a phenobarbital dose were excreted as the S– and R-enantiomers of the glucoside, respectively. Paibir et al.,56 working with human liver microsomes, found a S:R glucoside ratio of 6.75 ± SD 1.34:1.0. Even when N-glucoside formation is taken into consideration, a considerable discrepancy still exists between a phenobarbital intake and the measured output of the drug’s identified urinary excretion products. However, the N-glucosides of phenobarbital are pH-labile and begin to decompose once the environmental pH exceeds 5, forming barbiturate ring-opened derivatives.77 The subsequent fate of these ring-opened compounds is unknown. In vitro, at 37°C and a pH of 7.4, S-phenobarbital-N-glucoside has a half-life of about 15 hours.7 It is therefore possible that the phenobarbital-N-glucosides may undergo significant breakdown to as yet untraced derivatives while still present in the blood and tissues of the body, and also in urine while in the bladder. Hence N-glucosidation may possibly be the main elimination pathway for phenobarbital in humans.
Mephobarbital
Less than 3% of a mephobarbital dose is excreted in urine unmetabolized.20 The biotransformation of the R– and S-enantiomers of the drug is relatively stereoselective. The R-enantiomer is metabolized by CYP2C19 to yield phenolic derivatives;40 which are then transformed to glucuronides. The key role of CYP2C19 is evidenced by the much slower elimination of mephobarbital by subjects with the slow metabolizing alleles of the enzyme.39 Excretion of 4-hydroxyphenobarbital (p-hydroxyphenobarbital) glucuronide, predominantly as the R-enantiomer, accounts for some 35% of an oral dose of the racemic drug.33 It is likely that some of this 4-hydroxyphenobarbital is further metabolized to dihydrodiol and O-methyl catechol derivatives. A small amount of R-mephobarbital is oxidatively dealkylated to produce phenobarbital. [R]-mephobarbital may accumulate in plasma in poor metabolizers of the drug,34 although the extent of the biologic activity of this enantiomer is not known. In contrast, S-mephobarbital does not undergo aromatic hydroxylation, but is oxidatively dealkylated to yield phenobarbital, some of which is then further metabolized, as described earlier. Some 8% to 25% of a dose of R,S-mephobarbital appears in urine as phenobarbital,20 and phenobarbital formed from mephobarbital seems to account for most, if not all, of the drug’s biologic activity under steady-state conditions. It is not known whether N-glucosides are formed from either mephobarbital enantiomer.
Elimination
Phenobarbital is a low-clearance drug. Its total plasma clearance is about 0.004 L/kg per hour in adults, and declines in old age (0.0032 L/kg per hour in those over 65 years, as compared with 0.0041 L/kg per hour in adults aged 20–50 years [51]), but is up to 0.012 L/kg per hour in young children. Consequently phenobarbital has a relatively long half-life (3 to 5 days in adults and 1.5 days in children) that decreases with repeated administration of the drug,78 consistent with autoinduction of metabolism. Lim and Hooper,44 in six volunteers, all probably extensive metabolizers, showed that the clearance of S-mephobarbital was relatively low (0.017 ± SD 0.001 L/kg per hour) with an average half-life of 69.8 ± SD 14.8 hours. The oral clearance of R-mephobarbital was much higher (about
0.47 ± SD 0.18 L/kg per hour) and its half-life comparatively short (mean value, 7.5 ± SD 1.7 hours). Some evidence suggests that the clearance of R,S-mephobarbital is higher in the presence of enzyme-inducing agents.20 Details of the effects of age and sex on the disposition of the individual enantiomers of mephobarbital have been published.34
0.47 ± SD 0.18 L/kg per hour) and its half-life comparatively short (mean value, 7.5 ± SD 1.7 hours). Some evidence suggests that the clearance of R,S-mephobarbital is higher in the presence of enzyme-inducing agents.20 Details of the effects of age and sex on the disposition of the individual enantiomers of mephobarbital have been published.34
Because of its long elimination half-life, steady-state conditions may not be achieved until 2 to 3 weeks after a change in the dosage of phenobarbital. Although steady-state conditions would be reached earlier than this for S-mephobarbital when R,S-mephobarbital is used, the more clinically important consideration in mephobarbital use is the time when steady-state conditions also apply for the phenobarbital formed from the parent drug.
Concentration–Effect Relationships
Plasma phenobarbital concentrations may be used as a guide to dosing. Because of the long half-life of phenobarbital, under steady-state conditions, its plasma concentrations show very little fluctuation during interdose intervals of as long as 24 hours. Hence, steady-state predose measurement of the drug’s plasma concentration rarely offers advantages over measurements carried out at random stages during the dosage interval.
It has been proposed that plasma phenobarbital concentrations of 15 mg/L or higher would seem necessary to control seizures of a type likely to respond to the drug.22 However, target plasma phenobarbital concentrations recommended in the literature and employed in randomized clinical trials have ranged from 10 to 45 mg/L.9,19,30,50 There is also wide variation in the dosage required for seizure control as well as in that tolerated between individual patients due to various factors. For instance, evidence suggests that higher plasma concentrations might be needed to control partial than generalized tonic–clonic seizures.66 In general, plasma phenobarbital levels above 35 to 40 mg/L are increasingly likely to be associated with sedation.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


