Figure 60.1. Structural formulas of PRM and its main metabolites.
CHEMISTRY AND MECHANISM OF ACTION
Phenobarbital
Chemically, PB is 5-ethyl-5-phenylbarbituric acid (see Fig. 60.1). The molecular weight is 232.23, and the conversion factor from milligrams to micromoles is 4.31 (1 mg/L = 4.31 μmol/L). The sodium salt of PB is water soluble. PB in its free acid form is a white crystalline powder soluble in organic solvents, but with limited water and lipid solubility; it is a weak acid with a pKa of 7.3.
Many actions of PB at the cellular level have been described. Although it is not certain which are responsible for seizure protection, the available evidence seems to favor enhancement of γ-aminobutyric acid (GABA) inhibition (4). In animal models, PB protects against electroshock-induced seizures. Unlike phenytoin, carbamazepine, and PRM, PB also protects against seizures induced by chemical convulsants, such as pentylenetetrazol. In normal animals, PB raises the threshold and shortens the duration of afterdischarges elicited by electrical stimulation (5). Like other barbiturates, PB enhances postsynaptic GABAA receptor–mediated chloride (Cl−) currents by prolonging the opening of the Cl− ionophore (6). Increased flow of Cl− into the cell decreases excitability. Presynaptically, PB can cause a concentration-dependent reduction of calcium (Ca2+)-dependent action potentials (7), which may contribute to seizure protection at higher therapeutic levels and, especially, to sedative and anesthetic effects.
Primidone
Chemically, PRM is 5-ethyldihydro-5-phenyl-4,6(1-H,5H) pyrimidinedione. The molecular weight is 218.264, and the conversion factor from milligrams to micromoles is 4.59 (1 mg/L = 4.59 μmol/L). PRM is very poorly soluble in water, somewhat soluble in ethanol, and virtually insoluble in organic solvents.
The basic anticonvulsant action of PRM has been studied in mouse neurons in cell culture (8). PRM was compared with PB for its effect on amino acid responses and on sustained, high-frequency firing. In contrast to PB, PRM had no effect on postsynaptic GABA and glutamate responses at concentrations up to 50 μg/mL. However, both agents limited sustained, high-frequency, repetitive firing at relatively high concentrations (>50 μg/mL). Together, PRM and PB limited sustained high-frequency, repetitive firing at clinically relevant concentrations (12 and 20 μg/mL, respectively). The authors concluded that PRM and PB may act synergistically to reduce sustained, high-frequency, repetitive firing.
All the evidence regarding the individual antiepileptic properties of PRM and PB has been demonstrated in experiments with animals whose seizures were provoked (9–12). Because the metabolites accumulate a few hours after administration of the first dose, possible long-term protection by PRM alone against spontaneously occurring seizures cannot be assessed in humans. The anticonvulsant potency of PRM against maximal electroshock-induced seizures is similar to that of PB, but unlike PB, PRM was ineffective against chemically induced seizures caused by pentylenetetrazol or bicuculline (11). Thus, the experimental anticonvulsant spectrum of PRM differs from that of PB and is similar to that of carbamazepine and phenytoin. Therefore, PRM and PB may be two different AEDs with different mechanisms of action.
On the basis of brain concentrations in mice, PRM appears to be 2.5 times less neurotoxic than PB, with a superior therapeutic index (11). When PB and PRM were administered together in single-dose experiments in mice (13), their anticonvulsant activity was supra-additive (potentiated), and their neurotoxic effect was infra-additive. A PB–PRM brain concentration ratio of 1:1 provided the best therapeutic index. This ratio is not usually seen in patients, especially those taking PRM combined with enzyme-inducing drugs such as phenytoin or carbamazepine. If PRM is different from or even better than PB for the treatment of epilepsy, its different effect would be likely only when the PRM concentration equals or exceeds the PB concentration. Such a ratio is achieved only rarely with PRM monotherapy and almost never when PRM is added to phenytoin or carbamazepine, or combined with PB. The results of pharmacodynamic interactions between PRM and PB in mice were confirmed by experiments in amygdala-kindled rats. After single doses, the anticonvulsant effect of PB was potentiated by PRM, whereas side effects of PB, such as ataxia and muscle relaxation, were not increased by combined treatment with PRM (14).
In rats (15) and mice (11,12), PEMA had relatively weak anticonvulsant activity of its own. On the basis of brain concentrations in mice (11), PEMA was 16 times less potent than was PB in seizure protection and 8 times less potent in neurotoxic effects, but it potentiated the anticonvulsant (13,15) and neurotoxic effects (13) of PB. When this is interpreted together with the blood levels encountered in clinical practice, PEMA does not appear to significantly add to the antiepileptic effect or neurotoxicity of PRM therapy.
ABSORPTION, DISTRIBUTION, AND METABOLISM
Phenobarbital
Most formulations of PB contain sodium salt because of good aqueous solubility. The absolute bioavailability of oral preparations and intramuscular (IM) injection of PB is usually >90% (16,17). Accumulation half-life for the IM route (0.73 hours) was comparable to that to the oral route (0.64 hours). Time to peak concentration is usually 2 to 4 hours. In newborns, peak PB plasma levels after oral administration may be reached later than after IM administration (18). A parenteral solution of PB administered rectally has a bioavailability of 89%, compared with that of IM administration (19); average time to peak concentration was 4.4 hours.
PB is not highly bound to serum proteins (45%). Protein binding of PB is lower during pregnancy and in newborns, with a bound fraction between 30% and 40% in pregnant women and their infant offspring (20). Reported values for the volume of distribution vary. Following IV administration, average values were 0.54 L/kg in adult volunteers and 0.61 L/kg in adult patients with epilepsy (17), both well within the reported range. The volume of distribution of PB approached 1.0 L/kg in newborns (21).
PB is eliminated by both hepatic metabolism and renal excretion of the unchanged drug. An average of 20% to 25% of PB is eliminated unchanged by the kidneys in adults, with large interindividual variability (22,23). The two main metabolites of PB are p-hydroxyphenobarbital (PBOH) (see Fig. 60.1) and phenobarbital N-glucoside. At steady state, approximately 20% to 30% of the PB dose is transformed into PBOH by CYP2C9 (major), CYP2C19, and CYP2E1 (minor), approximately 50% of which is then conjugated to glucuronic acid (22–24). Nitrogen glucosidation, another relevant pathway of PB metabolism, accounts for 25% to 30% of total PB disposition (25). Other identified metabolites of PB represent a very low percentage of the total elimination.
The elimination of PB from serum follows first-order kinetics. The half-life of PB is age dependent. It is usually well above 100 hours in newborns (26) and averages 148 hours in asphyxiated newborns (27). During the neonatal period, PB elimination accelerates markedly; thereafter, half-lives are very short, with average values of 63 hours during the first year of life and 69 hours between the ages of 1 and 5 years (28). Half-lives in adults range between 80 and 100 hours, and no evidence of autoinduction of PB metabolism has been demonstrated (17).
Primidone
PRM is supplied as 50- and 250-mg tablets and as syrup (1 mL = 50 mg); extremely low solubility precludes parenteral administration. After oral ingestion of tablets, the time to peak serum concentrations in adult patients with epilepsy was 2.7 (29) and 3.2 hours (30), respectively, and 4 to 6 hours after single-dose administration in children (31). In the same study, an average of 92% of the dose (range, 72% to 123%) was excreted in the urine as unchanged PRM and metabolites, probably indicating complete oral bioavailability. Concomitant administration of acetazolamide reduced the oral absorption of PRM (32). One generic preparation was found to have a lower bioavailability than did the trademark product (33).
The volume of distribution of PRM ranged from 0.54 L/kg following acute intoxication (34) to 0.86 L/kg (35). The volume of distribution of PEMA after its oral administration was 0.69 L/kg (36). In human plasma, protein binding of both PRM and PEMA was <10% (15,30,36). Brain concentrations of PRM were found to be lower than simultaneous plasma concentrations in mice (11,12) and in rats (10). In patients undergoing surgery for intractable epilepsy, one group of investigators found an average brain-to-plasma ratio of 87% (37). In another report (12) of six patients whose mean plasma PRM concentration was 6.3 μg/mL, brain concentrations ranged between nondetectable and 2.2 μg/g. Brain concentrations of PEMA in mice ranged between 77% and 93% of the plasma levels (11,12). In humans, the cerebrospinal fluid–plasma ratio for PRM ranged from 0.8 to 1.13 (30,37,38), which is similar to human saliva-to-plasma ratios (39) and which is consistent with the high free fraction of plasma PRM.
The elimination half-life of PRM varies, mainly because of enzymatic induction by comedication. In adults receiving long-term PRM monotherapy, the elimination half-life ranged from 10 to 15 hours (40–42). Therapy with additional AEDs was associated with values of 6.5 and 8.3 hours (29,30,41,42). In 12 children (4 treated with PRM monotherapy, 8 treated with PRM and phenytoin), half-lives ranged from 4.5 to 11 hours (mean, 8.7 hours) (31). In newborns, however, the average PRM half-life was 23 hours (range, 8 to 80 hours) (43), which was associated with a limited biotransformation to the metabolites (44).
After oral ingestion of PEMA itself, the half-life of PRM was 15.7 hours (36). The elimination rate of PEMA cannot be determined accurately in patients taking PRM because the liver produces PEMA as long as PRM is measurable in the blood.
Because two metabolites of PRM accumulate after repeated administration of the agent and because both have independent anticonvulsant activity, an understanding of the qualitative and quantitative aspects of PRM metabolism is needed before any rational clinical use of this drug can be undertaken. Although relative efficacy and relative toxicity of PRM and its metabolites have been studied acutely in animals (11,13), similar investigations are virtually impossible in humans because the three compounds are always present simultaneously during long-term therapy.
Figure 60.1 shows the relevant metabolic pathways for PRM. The first metabolite of PRM to be identified, PEMA was found initially in rats (45) and thereafter in every species studied. PB and PBOH were discovered only 4 years later, in 1956 (46), and toxic reactions attributed to the derived PB were first reported in 1958 (47). Other metabolites of PRM, with either negligible or nondetectable blood levels during long-term therapy, have no practical significance. Numerous clinical studies have discussed the quantitative aspects of the biotransformation of PRM to PB and PEMA. A comparison of the ratios of PB serum levels to dose during long-term PB therapy and during long-term PRM therapy in the same patients demonstrated that 24.5% of the PRM dose is converted to PB (48). This is in accordance with the report that average PRM doses (in mg/kg/day) required to maintain a given PB level are about five times higher than the equivalent PB doses (49). The extent of PRM biotransformation and the ratios of the blood levels of PRM and its metabolites are very sensitive to interactions with other AEDs and are discussed separately. (See Table 60.1 for a summary of pharmacokinetics of PB and PRM)
INTERACTIONS WITH OTHER AGENTS
Phenobarbital
Most of the interactions of PB reflect its status as an enzymatic inducer that accelerates the biotransformation of some AEDs, as well as other agents. No clinically significant interaction with PB has been reported that involves absorption. Significant interactions involving displacement from serum proteins do not occur as PB is only 55% protein bound in serum. Clinically, the most significant interaction affecting PB levels is the inhibition of PB elimination by valproate (50). Seen in the majority of patients, the extent of this interaction is variable. As a result, the increase in PB concentration can reach 100%, often necessitating dosage adjustments. The concentrations of PB derived from PRM are equally affected by valproate.
In the great majority of interactions, PB affects levels of other agents. Levels of valproate (51) and carbamazepine (52) are often reduced by the addition of PB. Levels of the active metabolite of carbamazepine, the 10,11-epoxide, are less affected or may even increase, and the epoxide–carbamazepine ratio is usually higher in the presence of PB. Relative to the metabolism of phenytoin, PB appears to cause both enzymatic induction and competitive inhibition. The two effects tend to balance out in patients, and dosage adjustments of phenytoin are seldom necessary (53). PB significantly increases the clearance of lamotrigine (54), as well as that of ethosuximide, felbamate, topiramate, zonisamide, tiagabine (55), and rufinamide (56). Clobazam clearance does not appear to be affected by PB (57). Those AEDs that are predominantly cleared by renal elimination (e.g., levetiracetam, pregabalin, lacosamide) do not appear to interact with PB.
PB induces the metabolism of many agents besides AEDs, which may require closer monitoring and dosage adjustments. Some examples of drugs susceptible to this interaction include theophylline (58), warfarin (59), and steroids, including those contained in oral contraceptives, leading to breakthrough bleeding and contraceptive failure (60). Combined oral contraceptive preparations containing at least 50 μg of ethinyl estradiol have been recommended for some women taking PB (61,62). Of note, preparations with this higher content of ethinyl estradiol are currently indicated only for use in emergency contraception. This should only be explored when other methods of birth control are not feasible. Alternatives can be considered, such as intrauterine devices or medroxyprogesterone acetate injection, which is extensively metabolized on first pass through the liver, and concentrations are theoretically not affected by additional hepatic enzyme induction (63). However, as a conservative measure, the injection should be given more frequently (every 10 weeks instead of the usual 12 weeks) to ensure contraceptive efficacy. The use of the progesterone-only pill, progesterone implant, vaginal ring, and contraceptive patch are not recommended in women taking enzyme-inducing AEDs, unless combined with a barrier device. Regardless of which method is chosen, it is essential that the conversation between the patient and the practitioner, documenting a pregnancy or contraceptive plan, should appear frequently in the electronic medical records in women of childbearing potential.
Primidone
PRM is the cause, as well as the object, of numerous pharmacokinetic interactions (Table 60.1) (64). Because PB is invariably present during long-term PRM treatment, all of the effects of PB on other agents can be expected with PRM. The degree of enzymatic induction by other AEDs causes the extent of PRM biotransformation to vary among patients. Most reports describe enzymatic induction of the conversion of PRM to PB; some note inhibition. These interactions change not only the blood levels of PRM, PB, and PEMA relative to the PRM dose but also the ratios among the three substances. Phenytoin, a known potent inducer (41,65–67), causes the most extensive acceleration of PRM conversion, leading to a decrease in the PRM–PB serum concentration ratio. The rate of PRM biotransformation is slower with carbamazepine (41,64), which may also inhibit the conversion of PRM to PB, causing an increase in the PRM–PB serum concentration ratio (68). Table 60.2 summarizes the effect of comedication with phenytoin, carbamazepine, or both on the concentration-to-dose ratios and on the relative concentration ratios of PRM, PB, and PEMA (69). At the same daily dose of PRM, concomitant phenytoin or carbamazepine reduced the morning trough levels of PRM by about 50% and increased PB levels by about 160%. Thus, when patients receive concomitant phenytoin or carbamazepine, the average PRM dose required to maintain a given PB level is about 1.6 times lower than that with PRM monotherapy. Because derived PB is the product of enzymatic conversion and not the substrate, this difference is the opposite of what is usually seen with inducing interactions, which typically require an increased dose to maintain the same therapeutic drug level. With PRM, such an increase in metabolism often yields PB levels associated with toxic reactions.
Table 60.1 Pharmacokinetic Parameters of PB (17–21), PRM (29–31,34,35,40–42), and PEMA (36)
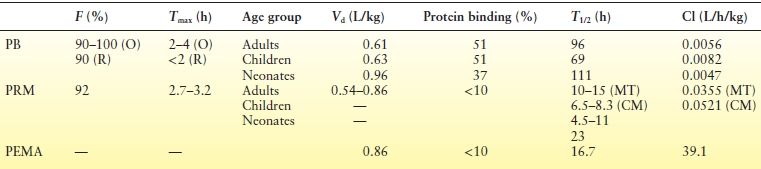
PB, phenobarbital; PRM, primidone; PEMA, phenylethylmalonamide.
O, oral; R, rectal; MT, monotherapy; CM, comedications.
Table 60.2 Serum Concentration: PRM Dose Ratios and Serum Concentration Ratios of PRM, PB, and PEMA at Steady Statea
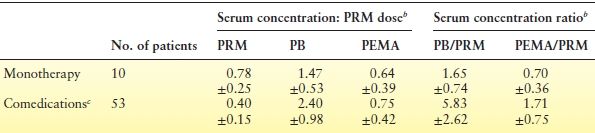
aAll blood samples were drawn before the first morning dose in hospitalized patients.
bMean ± standard deviation (SD), PRM dose in mg/kg/d, serum levels in mg/L.
cCombination therapy included phenytoin or carbamazepine, or both.
PRM, primidone; PB, phenobarbital; PEMA, phenylethylmalonamide.
From Bourgeois BFD. Primidone. In: Resor SR, Kutt H, eds. Medical Treatment of Epilepsy. New York: Marcel Dekker; 1992:371–378.
Table 60.2 also shows that the PB–PRM concentration ratio in a morning predose blood sample was more than three times higher in patients taking phenytoin or carbamazepine in addition to PRM (5.83 vs. 1.65, respectively). This means that at a PRM level of 10 mg/L, the corresponding average PB level would be 16.5 mg/L in a patient receiving PRM monotherapy, but 58.3 mg/L in a patient also taking phenytoin or carbamazepine.
Different effects of valproate on PRM kinetics have been described. In one study (70), transient elevations of PRM levels were observed after the addition of valproate; however, in general, no consistent changes in PRM or PB levels have been detected.
In all patients receiving long-term PRM therapy, the PB level is almost always higher than the PRM level. Attempts have been made to elevate the PRM level in relation to the PB level to obtain a greater therapeutic effect from PRM itself. Adding nicotinamide to the drug regimen (68) could achieve such a change in ratio, but the necessary doses may cause gastrointestinal side effects and hepatotoxic reactions. The antituberculosis drug isoniazid also markedly inhibits PRM biotransformation, producing relatively high PRM levels relative to PB levels (71).
EFFICACY
PB shows varying degree of efficacy against every seizure type, except absence seizures, but is used mainly for the treatment of generalized convulsive seizures and partial seizures. In a large-scale, controlled comparison of 622 adults with partial and secondarily generalized tonic–clonic seizures (72), phenytoin, carbamazepine, PB, and PRM were equally effective in achieving complete control. Compared to carbamazepine, PB and PRM controlled partial seizures in a lower percentage of patients. Evidence-based comparison of PB with phenytoin (73) and with carbamazepine (74) revealed no overall difference in seizure control, but PB was more likely to be withdrawn than the other two agents, presumably because of side effects. In children, PB was as effective as carbamazepine for up to 1 year in the treatment of partial seizures (39). In a randomized study of previously untreated children, PB, phenytoin, carbamazepine, and valproate were compared (75). After 6 of the first 10 children randomized to PB discontinued treatment mainly because of behavioral side effects, PB was eliminated from the study for ethical reasons. Generalized myoclonic seizures and, in particular, juvenile myoclonic epilepsy (76) also respond to PB, although it is not an agent of first choice.
As a major agent in the treatment of patients with convulsive status epilepticus, PB is a reasonable next-line option if seizures persist following administration of a benzodiazepine and phenytoin (77,78). In patients with status epilepticus, PB was as effective as a combination of diazepam and phenytoin (79). The main disadvantages associated with its use are respiratory depression and hypotension. Very high doses of PB have been recommended for the treatment of refractory status epilepticus in children (80,81). This approach controlled seizures when no limits were imposed relative to maximum dose, and serum levels of 70 to 344 mg/L were achieved (80). In this series, most patients were initially intubated but recovered good spontaneous respiration despite persistently high PB levels; hypotension was uncommon.
PB is the agent of first choice in newborns with any type of seizure, with control achieved in about one-third of the infants (21,82,83). An efficacy rate of 85% against various neonatal seizures was noted with loading doses of up to 40 mg/kg (84). In a recent study, newborns with seizures were randomized to initial treatment with PB or phenytoin (85). There was no difference in the percentage of neonates in whom seizure control was achieved with PB (43%) and with phenytoin (45%).
PB has been the most widely used agent for chronic prophylaxis of febrile seizures, with efficacy demonstrated at levels higher than 15 mg/L (86,87). Failure of prophylaxis was often due to noncompliance with the regimen and subtherapeutic levels at the time of seizure recurrence. However, such treatment is now rarely considered, for several reasons: improved understanding of the benign nature of simple febrile seizures; the efficacy of intermittent short-term use of rectal or oral diazepam therapy (88–90); and reservations about the possible detrimental effect on cognitive function (91,92).
Neurologic side effects have prevented PRM from becoming an agent of first choice for the treatment of any seizure type. Indications are similar to those for PB, except for the treatment of status epilepticus and neonatal seizures (PRM is not available in a parenteral formulation). PRM is effective against generalized tonic–clonic seizures and juvenile myoclonic epilepsy (93,94). However, other agents such as levetiracetam, lamotrigine, topiramate, zonisamide, and valproate have demonstrated efficacy and tolerability as first-line therapy (95) for the latter condition. The clinical efficacy of PRM and PB has been compared in various studies. Several demonstrated no superiority of PRM but did establish noninferiority (48,95,96). In one crossover study (97), the efficacy of PRM and PB was compared sequentially in the same patients. Similar PB levels were maintained during both therapies, and PRM was found to be slightly more effective than PB against generalized tonic–clonic seizures.
In partial and secondarily generalized seizures, PRM use was associated with the same degree of seizure control as PB, phenytoin or carbamazepine (98,99); however, the percentage of treatment failures was highest with PRM because of an increased incidence of side effects early on in treatment (72). PRM is rarely indicated for management of any type of seizure other than partial and secondarily generalized seizures. In particular, the agent has little or no place in the treatment of generalized epilepsies encountered in childhood, such as absence epilepsy and Lennox–Gastaut syndrome. Although some potential use has been demonstrated in the treatment of neonatal seizures (44), PRM is rarely used for this indication. PRM is contraindicated in any patient with a previous allergic, severe idiosyncratic reaction to PRM or to PB, and patients with hepatic porphyria. If indicated, the choice between PB and PRM may depend on individual factors. After PB has failed, PRM may still be tried. However, selecting PRM before PB may save one therapeutic step, based on the assumption that PB is unlikely to be effective if maximal tolerated doses of PRM have not controlled seizures.
ADVERSE EFFECTS
Among AEDs, PB and PRM are more likely to cause dose-related neurotoxic reactions, yet serious systemic side effects are rare. These agents invariably produce sedation and drowsiness at high doses in adults, whereas children often experience paradoxical behavioral side effects, mainly hyperactivity, aggressiveness, and insomnia, even at levels in the therapeutic range (<15 mg/L) (87). Sedation, usually present at relatively low levels during the first few days of treatment, subsides over time as tolerance to this effect develops. Sedation or somnolence reappears only at high therapeutic or supratherapeutic levels, usually >30 mg/L. As dose levels increase further, neurologic toxicity appears, characterized by dysarthria, ataxia, incoordination, and nystagmus. Movement disorders, such as dyskinesia, may be induced by PB, but they are rare (100). Like other AEDs, PB can exacerbate seizures or induce de novo seizures (101).
Depression has been attributed to both PB (102,103) and PRM therapy (104). Although its effect may have been overemphasized, double-blind, controlled studies have confirmed that PB affects cognitive abilities even at levels in the therapeutic range. Children treated with PB had lower memory and concentration scores than those receiving placebo, and these differences correlated significantly with plasma levels (105). Double-blind comparisons of PB-treated children versus untreated children (91,106) or valproate-treated children (107) demonstrated subtle but significantly lower intelligence quotient (IQ) scores in the PB groups. In an intention-to-treat analysis comparing children treated with PB or placebo for febrile seizures, the average IQ score was 8.4 points lower with PB (91) and remained 5.2 points lower 6 months after discontinuation of PB. Some differences persisted 3 to 5 years later (92).
Allergic rashes and hypersensitivity reactions are relatively rare with PB and PRM treatment (108), but cross-reactivity with CBZ and PHY has been established (109). Hematologic toxicity is quite rare with PB or with PRM (110,111). Like phenytoin and carbamazepine, PB can exacerbate acute intermittent porphyria (112) and cause osteoporosis, decreased bone mineral density, and increased risk of fracture, presumably through accelerated vitamin D metabolism (113–115). Vitamin K–deficient hemorrhagic disease in newborns of mothers treated with PB (116) can be prevented by administration of vitamin K to the mother before delivery. Connective tissue disorders associated with long-term PB therapy are well known (117) and have recently received renewed attention. These include Dupuytren contractures, plantar fibromatosis, heel and knuckle pads, frozen shoulder, and diffuse joint pain (118). Connective tissue disorders are an unusual side effect in children.
Like every AED, PB has been known to increase the risk for minor and major malformations in the offspring of mothers who were chronically exposed during pregnancy. Assessment of the specific risk for a given agent in clinical studies has been complicated by polytherapy and the underlying risk for malformation due to maternal epilepsy (119). A recent study using data from the North American AED Pregnancy Registry evaluated the relative safety of different AEDs used as monotherapy in pregnant women during the first trimester. PB was reported to be associated with a higher risk of major malformation when compared to lamotrigine (RR 2.9, 95% CI 1.4–5.8). It was also associated with a higher risk of cardiac defects and oral clefts (120). Like valproate and phenytoin, PB may be associated with reduced cognitive outcome in the child (121). Folic acid supplementation taken at 5 to 12 weeks of amenorrhea may decrease the risk of anomalies (122). Evidence that PB increases the risk for any type of tumor development in humans is lacking (123).
Acute and chronic toxic PRM reactions can be distinguished clearly from one another, but long-term PRM side effects are difficult to separate from those associated with derived PB. Because the ratio of PRM to PB varies, toxic side effects may occur at different PRM concentrations. Reliable evidence that long-term PRM side effects and potential teratogenic effects differ from those with comparable PB therapy is lacking. Ventriculoseptal defects, microcephaly, and poor somatic development (124) have been described in the offspring of women taking PRM, although no specific teratogenic pattern has been attributed to the agent.
The acute initial toxicity clearly differentiates PRM from PB. Even after a low initial dose of PRM, some patients experience transient side effects—usually drowsiness, dizziness, ataxia, nausea, and vomiting (71)—that are so debilitating they may be reluctant to take a second dose. Because this acute toxic reaction occurs before PB or PEMA is detected in the blood, it must be associated with PRM itself. Much larger doses of PRM are later tolerated by the same patients during long-term therapy, arguing for the development of tolerance to PRM probably within hours to days. The ratio of clinical toxicity score to serum PRM levels, determined in a group of patients receiving their first PRM dose (125), decreased significantly as early as 6 hours after the ingestion of drug. PB probably produces a cross-tolerance to this acute PRM toxicity and possibly to the anti-seizure activity, because patients on long-term PB therapy are less likely to experience the same degree of toxicity on first exposure to PRM (125–127).
CLINICAL USE
On the basis of its relative efficacy and toxicity profile, PB is no longer used as a first-line treatment for any seizure type, except for neonatal seizures. PB remains an agent of second or third choice for the treatment of generalized convulsive seizures and partial seizures at any age and is prescribed widely for infants because it is easier to use and is associated with less systemic toxicity than several other AEDs.
In adults, the daily maintenance dose of PB, between 1.5 and 4 mg/kg, achieves steady-state levels within the recommended therapeutic range of 15 to 40 mg/L. Because of its long elimination half-life and slow accumulation, the full maintenance dose can be administered on the first treatment day. Steady-state plasma level is attained only after 2 to 3 weeks. The daily maintenance dose of PB in children varies between 2 and 8 mg/kg; doses >8 mg/kg may be necessary in some infants to achieve high therapeutic levels. The dose is roughly inversely proportional to the child’s age: 2 months to 1 year, 4 to 11 mg/kg/day; 1 to 3 years, 3 to 7 mg/kg/day; and 3 to 6 years, 2 to 5 mg/kg/day (128). Given the long half-life of PB, dividing the daily dose of the agent into two or more doses appears unnecessary, even in children (129). Close monitoring of plasma levels and dosage reductions may be necessary in patients with advanced renal disease (130) and cirrhosis (131).
The IV loading dose of PB for the treatment of status epilepticus varies between 10 and 30 mg/kg; 15 to 20 mg/kg is most common. The rate of administration should not exceed 100 mg/min (2 mg/kg/min in children weighing <40 kg). PB penetrates the brain relatively slowly. Although full equilibrium is not reached for as long as 1 hour, therapeutic brain concentrations are reached within 3 minutes (132). The initial loading dose of 15 to 20 mg/kg in newborns is similar to the dose in children and adults and will achieve a plasma level of about 20 mg/L. This level can usually be maintained in newborns with a dose of 3 to 4 mg/kg/day (133). However, loading doses up to 40 mg/kg have been used in some situations (84).
PRM should be used alone or in combination with a noninducing drug, such as gabapentin, lamotrigine, topiramate, tiagabine, zonisamide, levetiracetam, vigabatrin, or a benzodiazepine. An inducing drug will shift the PRM–PB ratio to such an extent that the clinically effective component is solely PB. Similarly, prescribing PRM and PB simultaneously for the same patient is not justified. A low starting dose is more important with PRM than with most other AEDs because of the occurrence of transient, but severe, neurotoxic reactions. A first dose of one-half tablet (125 mg) at bedtime is often well-tolerated, but some patients initially need as little as one-quarter tablet (62.5 mg). The dose can then be increased every 3 days as tolerated, to a final daily maintenance dose of 10 to 20 mg/kg. Maintenance doses are 15 to 25 mg/kg/day in newborns, 10 to 25 mg/kg/day in infants, and 10 to 20 mg/kg/day in children.
A schedule that allows rapid advancement to the full maintenance dose of PRM was devised (134) based on the idea that PB produces cross-tolerance to the effects of PRM. After initial administration of PB, the dose is titrated as rapidly as tolerated to achieve a serum level up to 20 mg/L; abrupt switch to the full maintenance dose of PRM follows. Experimentation revealed that most adult patients tolerate the following regimen: 3 mg/kg of PB orally on day 1 (two doses of 1.5 mg/kg each, 12 hours apart); 3.5 mg/kg on day 2; 4 mg/kg on day 3; and 5 mg/kg on day 4 (Fig. 60.2). On day 5, the patient can receive a full PRM maintenance dose of 12.5 to 20 mg/kg, without significant toxicity. This beneficial effect of PB pretreatment on initial PRM toxicity has been confirmed in a more recent study (135).
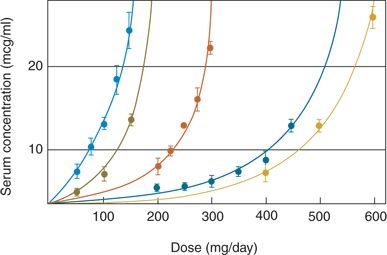
Figure 60.2. Phenobarbital (PB) loading dose over 4 days for rapid introduction of PRM. The PB values represent the average of 11 patients with standard deviation (vertical bars). The solid straight line connects the corresponding predicted values (5, 10, 15, and 20 mg/L). (Courtesy of Bourgeois BFD, unpublished data, 1991.)
Related posts:
 Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Continuous Spike-and-Wave During Sleep Including Landau–Kleffner Syndrome
Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Continuous Spike-and-Wave During Sleep Including Landau–Kleffner Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


