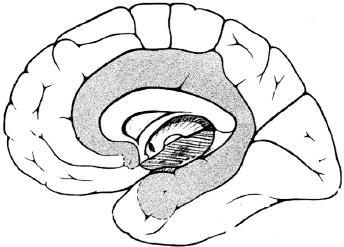
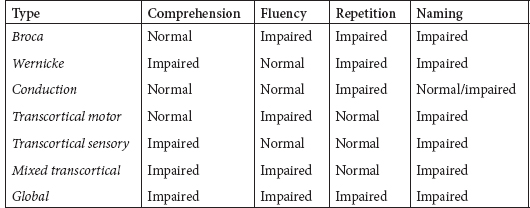
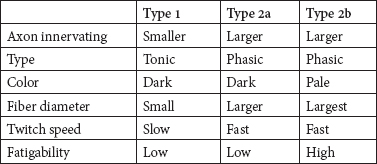
50 Practice Questions With Answers Practice Questions 1. Activation of a postsynaptic acetyl choline receptors results in which of the following? a. An efflux of Na+ into the cell and an efflux of K+, depolarizing the postsynaptic neuron b. An influx of Na+ into the cell and an efflux of K+, depolarizing the postsynaptic neuron c. An influx of Na+ into the cell and an influx of K+, depolarizing the postsynaptic neuron d. An influx of Na+ into the cell and an efflux of K+, repolarizing the postsynaptic neuron e. An efflux of Na+ into the cell and an influx of K+, repolarizing the postsynaptic neuron 2. Atropine and scopolamine a. block ACh actions only at muscarinic receptors. b. inhibit the release of AChr. c. prevent Ach receptor channel opening. d. prevent Ach receptor channel opening at motor end plate. e. block presymptomatic vesicle mobility. 3. Of the four main dopaminergic tracts, the a. nigrostriatal tract accounts for only a small part of the brain’s dopamine. b. tuberoinfundibular tract controls release of prolactin via D3 receptors. c. tuberoinfundibular tract controls release of prolactin via D2 receptors. d. mesolimbic tract controls release of prolactin via D3 receptors. e. mesocortical tract controls release of prolactin via D2 receptors. 4. Raphe nuclei a. project rostrally, mainly in the medulla and spinal cord. b. project caudally to the limbic structures and the cerebral cortex. c. project caudally, mainly to the medulla and spinal cord. d. stimulation produces effects similar to those of glutamate. e. lie dorsally in the diencephalon. 5. Which of the following is true regarding Prader-Willi syndrome and Angelman’s syndrome? a. Prader-Willi syndrome is paternally inherited with maternal imprinting; Angelman’s syndrome is paternally inherited with maternal imprinting. b. Prader-Willi syndrome is maternally inherited with maternal imprinting; Angelman’s syndrome is paternally inherited with paternal imprinting. c. Prader-Willi syndrome is maternally inherited with paternal imprinting: Angelman’s syndrome is paternally inherited with maternal imprinting. d. Prader-Willi syndrome is paternally inherited with maternal imprinting; Angelman’s syndrome is maternally inherited with paternal imprinting. e. In both Prader-Willi syndrome and Angelman’s syndrome, genes inherited from the father are silenced via methylation. 6. Trinucleotide repeats are seen in which of the following genetic conditions? a. Alzheimer’s disease; fragile X; Huntington’s disease b. Friedreich’s ataxia; AR torsion dystonia; myotonic dystrophy c. FTD; Segawa syndrome; fragile X d. Myotonic dystrophy; adult-onset focal dystonia; Alzheimer’s disease e. Fragile X; myotonic dystrophy; Friedreich’s ataxia 7. Which of the following clinical syndromes is matched with the appropriate region of protein/gene abnormality? a. Kallmann’s anosmia-hypogonadism: ATP-binding cassette transporter 7(ABCB7) b. Rett syndrome: cyclin-dependent kinase-like 5 (CDKL5) c. LIS1: reelin (RELN) d. LISX1: alpha-1-tubulin (TUBA1A) e. Holoprosencephaly: sonic hedgehog (SHH) 8. Which of the following statements related to disorders of primary neurulation is (are) correct? a. Spina bifida results from failure of the anterior neuropore to form. b. Spina bifida aperta is associated with a neurological deficit in 90% of those affected. c. Meningocele is a herniation of a cerebrospinal-fluid-filled sac with neural elements. d. Meningocele is a herniation of a cerebrospinal-fluid-filled sac without neural elements. e. B and D f. A, B, and C 9. Dandy-Walker syndrome is characterized by which of the following? a. Failure of foramen Magendie development; enlarged cerebellar vermis; agenesis of the corpus callosum b. Depression of the inion; cardiac abnormalities; increased migrational disorders c. Failure of foramen of Magendie development; cystic dilation of 4th ventricle; cerebellar vermis agenesis with enlarged posterior fossa d. Holoprosencephaly; elevation of the inion; agenesis of corpus callosum e. Absent septum pellucidum; hypoplastic optic nerves: schizencephaly 10. The shaded area represents which brain area? a. Somatosensory association cortex b. Parahippocampal gyrus c. Hippocampal gyrus d. Prefrontal cortex e. Limbic system 11. Which of the following thalamic nuclei is matched with the appropriate inputs and projections? a. Anterior: mammillary nucleus of the hypothalamus; cingulate gyrus b. Anterior: globus pallidus; striatum c. Pulvinar: association areas; striatum d. Ventroposterolateral: trigeminothalamic tracts; sensory cortex e. Lateral geniculate body: auditory input; primary auditory cortex 12. Which of the following superior-division middle cerebral artery branches is matched to the correct clinical syndrome? a. Ascending frontal branch: brachial monoplegia b. Ascending parietal: conduction aphasia c. Cortical-subcortical: brachial monoplegia d. Rolandic: primarily cortical sensory deficit e. Ascending parietal: agrammatic speech with normal comprehension 13. Which aphasia type is matched with the correct characteristics of comprehension, fluency, repetition, and naming? a. Transcortical sensory: spared comprehension, normal fluency, normal repetition, impaired naming b. Global aphasia: impaired comprehension, impaired fluency, spared repetition, impaired naming c. Broca: impaired comprehension, impaired fluency, impaired repetition, impaired naming d. Transcortical motor: spared comprehension, impaired fluency, normal repetition, impaired naming e. Conduction: spared comprehension, impaired fluency, normal repetition, impaired naming 14. Balint’s syndrome of oculomotor apraxia, optic ataxia, and asimultagnosia can be due to a. bilateral mesial frontal infarction. b. mesial hippocampal infarction. c. lateral temporal infarction. d. infarction of Striae of Gennari. e. bilateral occipito-parietal infarctions. 15. Contralateral paralysis of the arm and leg, contralateral impaired tactile and proprioceptive sense, and ipsilateral tongue paralysis are seen in which of the following syndromes? a. Posterior medullary region b. Avellis syndrome c. Wallenberg syndrome d. Jackson syndrome e. Medial medullary syndrome 16. IV thrombolysis with tissue plasminogen activator is approved by the FDA for which of the following? a. Ischemic stroke presenting within 4.5 hours b. Ischemic stroke presenting within 3 hours c. Hemorrhagic stroke presenting within 3 hours d. TIA presenting within 3 hours e. Lacunar stroke presenting within 3 hours 17. Concussion is defined as a. bruising of the brain without interruption of its architecture. b. contre-coup injury. c. violent shaking or jarring of the brain with transient functional impairment. d. head injury with definite loss of consciousness. e. focal injury at the site of impact. 18. Which of the following statements is correct regarding minor head injury? a. CT imaging is mandatory with minor head injury. b. All minor head injuries should be evaluated by a neurologist. c. There is a 1-in-1,000 chance of intracranial hemorrhage if no fracture and mentally clear. d. There is a 1-in-500 chance of intracranial hemorrhage if no fracture and mentally clear. e. There is a 1-in-100 chance of intracranial hemorrhage if no fracture and mentally clear. 19. Chronic subdural hematomas a. occur after a lucid period. b. generally occur weeks after a well-defined head injury. c. can cause aphasia as a common presentation. d. require immediate burr-hole evacuation. e. may be accompanied by giddiness, slowness of thinking, confusion, or apathy. 20. Which of the following is correct regarding treatment of refractory hypertension in the neurological ICU? a. Blood pressure should be reduced if SBP >185, regardless of baseline blood pressure. b. Nitroprusside is an acceptable treatment for hypertension in this setting. c. Nicardipine, hydralazine, or labetalol is recommended for the NICU setting. d. Aim is to keep MAP <90. e. Aim is to keep MAP <110. 21. Which of the following is true regarding intracranial hematoma expansion? a. It does not occur after first presentation except in rare circumstances. b. It is less likely in poorly controlled diabetics c. Patients may show the “spot sign.” d. Patients may show the “dot sign.” e. Antiplatelets increase the risk for ICH expansion. 22. Mild cognitive impairment (MCI) differs from dementia in that a. language disorders are uncommon in MCI. b. activities of daily living are generally spared. c. patient retains insight into problems with memory. d. MRI imaging differs between MCI and dementia. e. α beta 42 CSF levels are normal in MCI. 23. Alzheimer’s disease is a. more likely with higher education. b. more likely with moderate drinking. c. more likely with apolipoprotein E2 allele. d. less common with Down syndrome. e. more common with midlife obesity. 24. Which of the following is true regarding Lewy body disease (LBD) pathology? a. It consists of amyloid deposition in the striata nigra. b. Patient has antibodies against ubiquitin or alpha-synuclein. c. It never occurs with Alzheimer’s pathology. d. Patient has Lewy bodies (intracellular eosinophilic core with peripheral halo). e. Defects in NMDA have been described in LBD. 25. Frontotemporal dementia can present with which of the following? a. Semantic dementia; apathy; nonfluent aphasia b. ALS and behavioral disorders; cortical blindness; ataxia c. Nonfluent aphasia; memory disorder; myoclonic seizures d. Diffuse myoclonus; logopenic aphasia; simultanagnosia e. Depression; apraxia for limb kinetic activities; abulia 26. Migraine without aura includes a. headache attacks lasting 1 to 3 hours. b. improvement with exercise. c. bilateral temporal distribution. d. pulsatile quality. e. mild pain intensity. 27. A 54-year-old male patient comes to your practice. He has recurrent unilateral supraorbital attacks of pain lasting 2 minutes. These occur from 5 to 25 times a day. During the pain, he notices he has increased tearing. He has had these attacks for the past year and has had a workup for mass lesions, which was negative. You diagnose which of the following? a. Temporal arteritis b. Paroxysmal hemicranias c. Migraine with autonomic features d. Secondary headache due to undiagnosed orbital pseudotumor e. SUNCT 28. Which of the following is the most appropriate match between muscle fiber type and characteristics? a. Type 1: tonic; dark color; small fiber diameter; slow twitch speed; low fatigability b. Type 1: phasic; dark color; small fiber diameter; fast twitch speed; low fatigability c. Type 2a: phasic; pale color; largest fiber diameter; fast twitch speed; low fatigability d. Type 2b: Tonic; dark color; small fiber diameter; slow twitch speed; low fatigability e. Type 2b; Phasic; pale color; largest fiber diameter; slow twitch speed; high fatigability 29. In differentiating a peroneal nerve lesion from an L5 root lesion, which of the following signs is most useful? a. Knee flexion weakness b. Knee extension weakness c. Foot everter weakness d. Intrinsic foot muscle weakness e. Foot inverter weakness 30. Meralgia paresthetica a. is a psychiatric dissociation disorder. b. was described by Karl Jung. c. is due to entrapment of the lateral femoral cutaneous nerve. d. can be reliably diagnosed by electrophysiology. e. is usually treated surgically. 31. A 15-year-old boy comes into your clinic with his mother. He had an early morning generalized tonic-clonic seizure 2 weeks ago and is now on phenytoin 100 mg three times a day. His mother notes that in the mornings, he may have episodes of staring for a few seconds, and tends to jerk his arms for an hour after awakening. There is no developmental or family history of significance. His EEG shows 4- to 5-Hz generalized spike and wave. The mother notes that the jerks and staring spells are more prominent since beginning the medicine. Your next move is to a. raise the phenytoin to 200 mg twice a day b. switch to carbamazepine. c. switch to lacosamide. d. switch to valproic acid. e. switch to topiramate. 32. A 1-year-old child is brought to your clinic by his parents. At age 4 months he began to have episodes of suddenly flexing his body and arms. Development has slowed, and he has episodes hundreds of times a day. His EEG shows hypsarrhythmia. Imaging has not yet been done. Your diagnosis is which of the following? a. Aicardi’s syndrome b. Lennox-Gastaut syndrome c. MERRF d. Sialidosis type 1 e. West’s syndrome 33. A 23-year-old female known to have complex partial seizures treated with carbamazepine is brought to the emergency room after having had three seizures in the past half hour. She remains unconscious, breathing shallowly. After ABCs, insertion of an IV, and institution of monitoring, you decide to treat her by a. giving diazepam 10 mg IV. b. giving pentobarbital due to refractory status. c. giving midazolam due to refractory status. d. loading IV carbamazepine. e. giving lorazepam 0.1-mg/kg bolus. 34-38. Match each movement disorder with the appropriate MRI finding. 34. PKAN a. “Hot cross bun” sign 35. PSP b. “Eye of the tiger” sign 36. MSA c. Hummingbird sign 37. CBD d. Asymmetric MRI atrophy 38. Variant CJD e. Pulvinar sign 39. You have a 34-year-old patient with relapsing-remitting MS who has had three relapses treated with high-dose IV solumedrol in the past year. Her MRI continues to show new lesion activity with gadolinium-positive lesions despite treatment with a low-dose interferon. Your next treatment strategy might include the following, but will be tempered by the risk of the linked complication: a. Natalizumab; treatment-related acute leukemia b. Fingolimod; autoimmune hepatitis c. Alemtuzumab; idiopathic thrombocytopenic purpura d. Cyclophosphamide; Grave’s disease e. Mitoxantrone: progressive multifocal leukoencephalopathy 40. You have a 12-year-old patient who, after an initial viral illness, develops confusion, fever and tachycardia, nystagmus, brisk reflexes, pale optic discs, and upgoing toes. The MRI shows diffuse white-matter lesions that also affect the basal ganglia and spinal cord. CSF shows 34 white cells 90% lymphs, protein slightly elevated, and normal CSF glucose with no oligoclonal bands. You make which diagnosis? a. Acute necrotizing hemorrhagic encephalomyelitis b. Schilder’s disease c. Neuromyelitis optica spectrum disorder d. Acute disseminated encephalomyelitis e. Baló’s concentric sclerosis 41. West Nile virus infection a. usually causes a Guillain-Barre-like illness. b. causes symptoms in most affected individuals. c. occurs primarily in young, healthy teenagers. d. shows characteristic lesions on MRI. e. rarely causes a syndrome of acute flaccid paralysis. 42. A 16-year-old male develops behavioral and personality changes followed 1 month later by myoclonus, ataxia, and difficulty with language. After 3 months the patient develops visual loss and quadriparesis, and gradually slips into a coma. Late in his course the EEG shows burst suppression. This condition a. is a delayed response to herpes zoster infection. b. is due to a slow virus infection. c. can be effectively treated by rituximab. d. is due to a rubella. e. is due to defective viral maturation in the brain. 43–46. Match each neurotoxin to the appropriate clinical finding. 43. Arsenic poisoning a. Alopecia 44. Thallium poisoning b. Mees’ lines 45. Lead poisoning c. Bradykinesia 46. Manganese poisoning d. Burtonian line 47. Triphasic waves a. are pathognomonic of hepatic encephalopathy. b. are pathognomonic for CJD. c. are a late finding of status epilepticus. d. are seen in a variety of encephalopathies. e. respond to benzodiazepine treatment. 48. Which of the following is true regarding the hypogastric nerve? a. It provides striated muscle relaxation via beta-adrenergic receptors. b. The postganglionic efferent nerves exit at T10-L2. c. The nerves travel via the hypogastric nerve to the superior pelvic plexus. d. It provides smooth muscle relaxation via alpha-adrenergic receptors. e. It provides smooth muscle relaxation via beta-adrenergic receptors. 49. Downbeat nystagmus is a sign of which of the following? a. A lesion of the parasellar region b. A lesion of the dorsal midbrain c. Lesion of Mollaret’s triangle d. A massive pontine lesion e. A lesion of the cervicomedullary junction 50. Ménière’s syndrome a. causes persistent hearing loss from onset. b. causes positional vertigo. c. is due to reduction of endolymph pressure. d. causes episodic vertigo. e. causes vertigo with compression of the tragus. And 10 More Bonus Questions 1. Which of the following is true regarding SIADH? a. Laboratory findings show hyponatremia with inappropriate urine osmolality. b. Laboratory findings show hypernatremia with inappropriate urine osmolality. c. Laboratory findings show euvolemia with inappropriate urine osmolality. d. It is improved by increased free water. e. It is often seen after severe head injury. 2. Which of the following is true regarding oligodendrogliomas? a. They account for 25% of intracranial gliomas. b. They usually affect the brainstem. c. Deletion of 1p19q causes a better treatment response. d. They rarely calcify. e. They cause a scrambled-egg appearance on pathology. 3–6. Match each syndrome with the most common antibody syndrome. 3. Progressive encephalomyelitis a. GAD-65 4. Sensory neuropathy b. anti-Hu 5. Stiff-person syndrome c. VGCC 6. Lambert-Eaton syndrome d. GlyR 7. Features related to clozapine include which of the following? a. Lack of EPS; agranulocytosis; elevated seizure threshold b. More dopamine than 5-HT blockade c. Approved for treatment of TD: most effective antipsychotic d. Mild anticholinergic effects; lowers seizure threshold e. Agranulocytosis: primarily D3 blockade 8. Fragile X syndrome is caused by a a. binucleotide repeat at chromosomal locus Xq27.3. b. trinucleotide repeat at chromosomal locus Xq27.3. c. trinucleotide repeat at chromosomal locus Xq26.3. d. tetranucleotide repeat at chromosomal locus Xq26.3. e. trinucleotide repeat at chromosomal locus Xq25.3. 9. Anton’s syndrome causes a. right homonymous hemianopsia. b. apraxia of the left hand. c. retention of reactive pupils. d. prosopagnosia. e. visuospatial disorientation. 10. Klüver-Bucy syndrome a. is usually seen with posterior temporal damage. b. may be seen with unilateral temporal damage. c. is associated with hyposexuality. d. is associated with hyperorality. e. results in inattention to stimuli in the environment. Practice Answers 1. b. An influx of Na+ into the cell and an efflux of K+, depolarizing the postsynaptic neuron The voltage-gated calcium channel is open as the action potential (AP) reaches the terminal bouton of the presynaptic neuron, producing an influx of calcium ions that allows exocytosis of presynaptic vesicles containing ACh into the synaptic cleft. The activation of postsynaptic ACh receptors results in an influx of Na+ into the cell and an efflux of K+, which depolarizes the postsynaptic neuron, propagating a new AP. 2. a. Block ACh actions only at muscarinic receptors. Atropine and scopolamine block ACh actions only at muscarinic receptors. Botulinum toxin inhibits the release of ACh. Beta bungarotoxin prevents ACh receptor channel opening. D-Tubocurarine prevents ACh receptor channel opening at motor endplates. Botulinum toxin blocks presymptomatic vesicle mobility. 3. c. tuberoinfundibular tract controls release of prolactin via D2 receptors. The nigrostriatal tract accounts for most of the brain’s dopamine. The tuberoinfundibular tract controls release of prolactin via D2 receptors. 4. c. project caudally, mainly to the medulla and spinal cord. The raphe nuclei also project rostrally to the limbic structures and cerebral cortex. Stimulation of the raphe nuclei produces effects similar to those of lysergic acid diethylamide (LSD). Raphe nuclei are located in the brainstem. 5. d. Prader-Willi syndrome is paternally inherited with maternal imprinting; Angelman’s syndrome is maternally inherited with paternal imprinting. DNA and histone methylation allow transcriptional silencing of individual genes, as well as regions of chromosomes. Genomic imprinting is the regulation of gene expression depending on the parental origin of the gene. In paternal imprinting, genes inherited from the father are silenced through methylation, and only those from the mother are expressed. Maternal imprinting is the opposite. 6. e. Fragile X; myotonic dystrophy; Friedreich’s ataxia CAG repeat diseases are the most common trinucleotide repeat syndromes in neurology. Examples include Huntington’s, spinocerebellar ataxia type 1 and other SCAs, spinobulbar muscular atrophy, and dentatorubropallidoluysian syndrome. Fragile X is a CGG repeat (“Child with Giant Gonads”). Myotonic dystrophy is due to a CTG repeat (“Continues To Grasp”). Friedreich’s ataxia is due to a GAA repeat (“German [name] AtaxiA”). 7. e. Holoprosencephaly: sonic hedgehog (SHH) Kallmann’s syndrome (anosmia, hypogonadism) is an X-linked recessive disorder related to abnormalities in the enzyme anosmin related to the gene KAL1. Rett syndrome is an X-linked dominant condition related to abnormalities in methyl-CpG-binding protein 2 (gene MECP2). LIS1 (Miller-Dieker syndrome) is autosomal recessive and related to abnormalities in the enzyme LIS1 (gene PAFAH1B1). LISX1 is an X-linked dominant condition related to abnormalities of doublcortin (gene DBX). 8. e. B and D Spina bifida aperta is associated with a neurological deficit in 90% of cases. Meningocele is a herniation of a cerebrospinal-fluid-filled sac without neural elements. Spina bifida results from failure of the posterior neuropore to form. Myelomeningocele: herniated neural elements covered by meningeal sac; 80% are lumbar; 90% have hydrocephalus if lumbar is involved; symptoms include motor, sensory, and sphincter dysfunction. Myeloschisis: neural elements at surface completely uncovered; associated with malformed skull base; most babies are stillborn. Myelocystocele: herniation of meninges and cord with dilated central canal. 9. c. Failure of foramen of Magendie development; cystic dilation of 4th ventricle; cerebellar vermis agenesis with enlarged posterior fossa Dandy-Walker malformation is association with failure of foramen of Magendie development; cystic dilation of 4th ventricle; cerebellar vermis agenesis with enlarged posterior fossa; elevation of the inion; agenesis of the corpus callosum; 70% with migrational disorders; associated with cardiac abnormalities and urinary tract infections; frequency: 1:25,000; may result from riboflavin inhibitors, posterior fossa trauma, or viral infection. Septo-optic dysplasia is associated with absent or hypoplastic septum pellucidum, hypoplastic optic nerves, schizencephaly in approximately 50% but normal-sized ventricles, pituitary axis dysfunction (50% with diabetes insipidus). 10. e. Limbic system The limbic system incorporates several structures involved in emotion, memory, olfaction, and other evolutionarily ancient functions. The limbic pathways include the circuit of Papez: subiculum–fornix–mamillary body–mamillothalamic tract–anterior nucleus of thalamus–anterior limb of internal capsule–cingulate gyrus–cingulum–entorhinal cortex–perforant pathway–subiculum and hippocampus; olfactory projections; hippocampal formation projections and amygdalar connections. 11. a. Anterior: mammillary nucleus of the hypothalamus; cingulate gyrus The anterior thalamic nuclei have inputs from mammillary nuclei of the hypothalamus and projections to the cingulate gyrus. The pulvinar connects reciprocally with large association areas of the parietal, temporal, and occipital lobes. The ventroposterolateral nuclei have inputs from the spinothalamic tracts and medial lemnisci, and project to the sensory cortex. The ventroposteromedial nuclei receive input from trigeminothalamic tracts and nucleus solitarius and project to the sensory cortex. The lateral geniculate body has inputs from the retina via the optic tract, and projects to the visual cortex. The medial geniculate body receives auditory input via the brachium of the inferior colliculus and projects to the primary auditory cortex. 12. c. Cortical-subcortical: brachial monoplegia The cortical-subcortical branches of the superior division of the MCA cause a brachial monoplegia. Ascending frontal branches cause initial mutism and mild comprehension defect, then slightly dysfluent, agrammatic speech with normal comprehension. Rolandic branches are associated with sensorimotor paresis with severe dysarthria but little aphasia. Ascending parietal: no sensorimotor defect, only a conduction aphasia. 13. d. Transcortical motor: spared comprehension, impaired fluency, normal repetition, impaired naming 14. e. bilateral occipito-parietal infarctions. Balint’s syndrome is usually caused by watershed infarctions between the posterior and middle cerebral artery territories caused by hypoperfusion. It causes oculomotor apraxia (inability to direct eyes to object of interest), optic ataxia (failure to grasp objects under visual guidance), and simultanagnosia (inability to perceive more than a single object at a time in a scene that contains more than one object). 15. e. Medial medullary syndrome Lateral medullary syndrome/Wallenberg syndrome: vestibular nuclei (nystagmus, oscillopsia, vertigo, nausea, vomiting); spinothalamic tract (contralateral impairment of pain and thermal sense over one-half the body); descending sympathetic tract (ipsilateral Horner’s—ptosis, miosis, anhidrosis); cranial nerves (CNs) IX and X (hoarseness, dysphagia, ipsilateral paralysis of palate and vocal cord, diminished gag); otolithic nucleus (vertical diplopia and illusion of tilting of vision); olivocerebellar and/or spinocerebellar fibers/restiform body (ipsilateral ataxia of limbs, falling to ipsilateral side); nucleus and tractus solitarius (loss of taste); descending tract and nucleus of V (pain, burning, impaired sensation on ipsilateral one-half of face; rarely nucleus cuneatus and gracilis (ipsilateral numbness of limbs); most likely due to occlusion of vertebral artery (eight-tenths) or posterior-inferior cerebellar artery. Opalski syndrome: considered a variant of lateral medullary syndrome with ipsilateral hemiplegia, likely due to caudal extension of the infarct due to involvement of perforator branches arising from the distal vertebral artery. Medial medullary syndrome: involves medullary pyramid (contralateral paralysis of arm and leg); medial lemniscus (contralateral impaired tactile and proprioceptive sense over one-half the body); CN XII (ipsilateral paralysis and, later, hemiatrophy of the tongue). Hemimedullary infarction (Babinski–Nageotte syndrome): occlusion of the ipsilateral vertebral artery proximal to the posterior-inferior cerebellar artery and its anterior spinal artery causes medial medullary syndrome and lateral medullary syndrome simultaneously. Posterior medullary region: ipsilateral cerebellar ataxia and, rarely, hiccups. Avellis syndrome: tegmentum of medulla: CN X, spinothalamic tract (paralysis of soft palate and vocal cord and contralateral hemianesthesia). Jackson syndrome: tegmentum of medulla: CN X, XII, corticospinal tract (Avellis syndrome plus ipsilateral tongue paralysis). 16. b. Ischemic stroke presenting within 3 hours The NINDS Tissue Plasminogen Activator (tPA) trial established the efficacy of intravenous (IV) tPA for patients presenting within 3 hours from symptom onset. Compared to placebo, patients treated with tPA had a 16% absolute increase in favorable outcomes at 3 months. IV tPA is approved by the FDA for ischemic stroke presenting within 3 hours from last known normal. No consent is required for this time window. For patients presenting after 3 hours but before 4.5 hrs, ECASS III was the first study to show a statistical benefit for IV tPA use, with a slightly higher intracranial hemorrhage rate. The American Heart Association and American Stroke Association recommend the use of IV tPA for patients presenting between 3 and 4.5 hours from last known normal; however, this is not FDA approved. 17. c. violent shaking or jarring of the brain with resulting transient functional impairment. Loss of consciousness is not required for the diagnosis of concussion. Contusion is bruising of the brain without interruption of its architecture. A coup injury usually occurs when the head is immobilized, and damage is focused at the site of impact. A contre-coup injury is injury opposite to the site of impact due to the head not being immobilized (brain thrown into opposite region of the skull). 18. c. There is a 1-in-1,000 chance of intracranial hemorrhage if no fracture and mentally clear. CT imaging is not mandatory with minor head injury. Neurologists are not needed in the evaluation of all minor head injuries. 19. e. May be accompanied by giddiness, slowness of thinking, confusion, or apathy Chronic subdural hematomas are generally due to traumatic injury that was trivial or forgotten. A period of weeks passes before onset of headaches, giddiness, slowness of thinking, confusion, apathy, drowsiness, or seizures. CSF may be clear, bloody, acellular, to xanthochromic (generally not recommended due to mass effect; diagnosis usually by CT or MRI). A subdural hygroma (collection of blood and CSF in subdural space) may form in the potential space created by the SDH. 20. c. Nicardipine, hydralazine, and labetalol are recommended for the NICU setting. Blood pressure reduction should take into account prehospitalization baseline blood pressures. Nitroprusside may cause venodilation, which may lead to increased intracranial pressure. Aim is to keep MAP (mean arterial pressure) less than 130. 21. c. Patients may show the “spot sign.” CT may show a hyperdense spot within a hematoma, suggesting active bleeding. CTA may assist in locating a bleeding source. Nearly half of all ICHs expand in size from presentation within first few hours. Most stabilize in 24 hours. Most ICH patients should be monitored in an appropriate ICU. Antiplatelets have not been shown to increase risk for ICH expansion. 22. b. activities of daily living are generally spared. Patients with MCI can have language disorders, although usually they present with memory impairment. Patients may not retain insight into memory problems, although issues with insight tend to be less than with dementia. MRI imaging does not discriminate between MCI and dementia. α-beta 42 CSF levels may be reduced in MCI prior to onset of dementia. They may predict Alzheimer’s pathology but do not indicate present severity of cognitive deficit. 23. e. more common with midlife obesity. Alzheimer’s disease is the most common degenerative disease of the brain. The incidence increases sharply with age after 65. Age is the most important risk factor. Other risk factors include Down syndrome (patient 30–45 y/o shows similar pathologic changes), midlife obesity, diabetes mellitus, current tobacco use, head injury, apolipoprotein E 4 genotype; reported protective factors: education, Mediterranean-type diet, low or moderate alcohol intake, physical activity, inheritance of apolipoprotein E2 allele. Dominant AD can occur with multiple genetic mutations (presenilin 1 [PS1] most common, presenilin 2 [PS2], amyloid precursor protein [APP], C9 open reading frame 72, Trem2, etc.). 24. b. Patient has antibodies against ubiquitin or alpha-synuclein. LBD shows Lewy bodies in brainstem nuclei, subcortical regions, and cerebral cortices. In the brainstem, pigmented neurons often present with the classic morphology of intracellular LBs, comprising an eosinophilic core with a peripheral halo; immunohistochemistry using antibodies against ubiquitin or α-synuclein has been shown to be more sensitive and specific in the detection of cortical LB; the clinical overlap of AD, DLB, and PD with dementia similarly extends to their pathology—most cases of DLB have varying degrees of AD pathology, including deposits of β-amyloid protein and neurofibrillary tangles. Neurochemistry: substantial loss of cholinergic neurons in the nucleus basalis of Meynert, suggesting a cholinergic mechanism of cognitive impairment in DLB, similar to that of AD; deficits in γ-aminobutyric acid (GABA), dopamine, and serotonin neurotransmission have also been described in DLB; neocortical choline acetyltransferase, a synthetic enzyme for acetylcholine, is decreased significantly, similar to that seen in AD or PD with dementia; reduced dopamine and its metabolites have been shown in DLB brains, possibly accounting for its parkinsonian features. 25. a. Semantic dementia; apathy; nonfluent aphasia Frontotemporal dementia can have various presentations. Most commonly presents with behavioral variant, either disinhibition or apathy, often with personality change, loss of judgment, altered appetites (often bizarre appetites), difficulty with decision making, and sometimes sexual disinhibition. Other variants include semantic dementia, with problems with fluent empty speech, naming impairment, and difficulty with identifying objects. Occasionally patients have a progressive nonfluent aphasia. Some patients can have ALS and FTD in combination. 26. d. pulsatile quality, Migraine without aura: At least five attacks fulfilling criteria Headache attacks lasting 4 to 72 hours Headache has at least two of the following characteristics: Unilateral location Pulsating quality Moderate or severe pain intensity Aggravation by or causing avoidance of routine physical activity During headache at least one of the following: Nausea and/or vomiting Photophobia and phonophobia Not attributed to another disorder 27. e. SUNCT SUNCT (short-lasting unilateral neuralgiform headache with conjunctival injection and tearing) is a rare headache syndrome, most common men over 50, involving bursts of moderate to severe burning, piercing, throbbing pain around eye or temple one side. Peaks in seconds lasts from 5 seconds to 4 minutes. Associated with conjunctival injection, nasal congestion, runny nose, sweaty forehead, swelling of the eyelid, and pressure in the eye. May occur up to 5 to 6 times per hour. Corticosteroids, gabapentin, lamotrigine, and carbamazepine may be helpful. Glycerol injections have been used in severe cases. Temporal arteritis causes persistent headache without autonomic features. Paroxysmal hemicrania is a rare adult-onset headache syndrome. Pain is severe throbbing or boring pain on one side of face. Associated with red and tearing eyes, drooping eyelid. Episodes occur 5 to 30 times per day and last 2 to 30 minutes. NSAIDs, particularly indomethacin, provide relief, but dosing may need to be high. Migraines last longer than this headache syndrome. Orbital pseudotumor has been ruled out. 28. a. Type 1: tonic; dark color; small fiber diameter; slow twitch speed; low fatigability Types of muscle fibers 29. e. Foot inverter weakness Everters and dorsiflexors of the foot are innervated by peroneal nerve muscles. Foot inverters are innervated by the tibial nerve. Both are partially supplied by L5. Thus in a peroneal nerve palsy, foot inverters will be spared, but will be weak in L5 root disorder. 30. c. is due to entrapment of the lateral femoral cutaneous nerve. Meralgia paresthetica is an entrapment of the lateral femoral cutaneous nerve, usually as it enters the pelvis between the inguinal ligament and its attachment to the anterior superior iliac spine. It was described by Sigmund Freud in the 1800s. It cannot be reliably diagnosed by electrophysiology in many cases. It is usually treated conservatively with looser garments and belts, weight reduction, reposition of seats, and possibly medications to reduce burning paresthesias. 31. d. switch to valproic acid. This case is characteristic of juvenile myoclonic epilepsy. This subtype of myoclonic epilepsy usually begins at age 12 to 16 with myoclonic events and tonic-clonic seizures with occasional atypical absence. Genetic localization is chromosome 6p. May be increased by photic stimulation and sleep deprivation. The treatment of choice is valproic acid, and patients may not respond to other, older antiepileptics. 32. e. West’s syndrome This patient has typical age of onset and clinical manifestations of West’s syndrome. This begins between age 3 months and 3 years, with tonic spasms (brief, rapid tonic contractures of trunk and limbs lasting few seconds), which may be flexor, extensor, or mixed. These may occur up to hundreds of times a day. Patients experience mental retardation. EEG shows hypsarrhythmia, a characteristic pattern with chaotic high- to extremely-high-voltage delta and theta with superimposed multiple spike and wave. Treatment consists of ACTH or vigabatrin. Aicardi’s syndrome is an X-linked disorder with onset at birth presenting with infantile spasms, hemiconvulsions, agenesis of the corpus callosum, coloboma, and vertebral anomalies. MERRF (mitochondrial epilepsy with ragged red fibers) is a mitochondrial disorder with onset at age 3 to 65 that presents with myoclonic epilepsy, cerebellar dysfunction, and other features suggestive of a mitochondrial disorder. Sialidosis I is an autosomal recessive disorder due to decreased alpha-neuraminidase. Onset is in adolescence, with severe myoclonus, visual impairment, and cherry-red spots in the fundi. Lennox-Gastaut is a disorder with an onset of age 1 to 10 years presenting with multiple seizure types, with slow spike and wave pattern. 33. e. giving lorazepam 0.1-mg/kg bolus. After evaluating and treating ABCs, the next step in the treatment of status epilepticus is giving lorazepam 4 to 10 mg IV (0.1-mg/kg bolus), as this is the most rapid and effective treatment for status epilepticus. Fosphenytoin may be given after this. Carbamazepine is not available IV. Pentobarbital and midazolam have been used for refractory status, but this patient does not meet this criterion. Diazepam redistributes to fatty tissues within minutes, so is not an optimal therapy for initial status treatment. 34. b 35. c 36. a 37. d 38. e 34–38. Pantothenate kinase-associated neurodegeneration (PKAN) was formerly known as Hallervorden-Spatz syndrome; it is one of the rare neurodegeneration disorders with brain iron accumulation. Imaging shows decreased T2-weighted signal in the globus pallidus and substantia nigra, sometimes accompanied by hyperintense area within the hypodense zone (“eye of the tiger”). Progressive supranuclear palsy (PSP) is a progressive neurodegenerative disorder primarily affecting the midbrain and causing toppling gait, dysarthria, vertical gaze disorder, and other findings. MRI may show thinning of the midbrain, causing a “hummingbird sign” on sagittal imaging. Multiple-system atrophy (MSA) is a degenerative disorder affecting multiple systems. It may show a sign of crisscross linear atrophy on MRI of the pons, the so-called “hot cross bun” sign. Corticobasal degeneration (CBD) is a group of disorders that manifest with focal cortical symptoms such as apraxia and visuospatial deficits, dystonic limb with stimulus sensitive myoclonus, and cortical sensory findings. Focal brain atrophy in parietal regions on MRI may accompany this. Creutzfeldt-Jakob disorder (CJD) is a rapidly progressive prion disorder. On imaging, variant CJD may show the “pulvinar sign” with altered signal in the dorsal thalamus. 39. c. Alemtuzumab; idiopathic thrombocytopenic purpura Natalizumab, a monoclonal that blocks lymphocyte entry into the CNS, increases the risk of progressive multifocal leukoencephalopathy, particularly when patients are positive for John Cunningham virus antibody (JCVAB +). Fingolimod binds to the SIP receptor of the lymphocyte, internalizing the receptor and interfering with the exit of lymphocytes from the lymph nodes. Risks include first-dose bradycardia, disseminated zoster infections, and macular edema. Alemtuzumab is a monoclonal antibody that depletes B and T cells for months after initial treatment. It can cause Grave’s disease or ITP due to return of certain B-cell lines predisposing to immune disorders. Cyclophosphamide is a chemotherapy that can cause immunosuppression and hair loss. Mitoxantrone is a chemotherapy that can cause treatment-related acute leukemia and cardiomyopathy. 40. d. Acute disseminated encephalomyelitis ADEM is an autoimmune demyelinating disorder usually seen in children after a viral illness or a vaccination. It is acute, usually with encephalopathy and systemic symptoms, with diffuse white-matter lesions in the spinal cord and brain that can include basal ganglia and brainstem. Schilder’s disease is a poorly characterized demyelinating disorder of children; it had a progressive course leading to death with elevated CSF myelin basic protein. Neuromyelitis optica spectrum disorders are caused by antibodies directed at aquaporin-4 located at astrocytic foot processes. Course is relapsing primarily with optic neuritis or myelitis, but occasionally with atypical brain events such as refractory vomiting due to lesions near the fourth ventricle. Acute necrotizing hemorrhagic encephalomyelitis is a rare fulminant form of demyelination with hemorrhagic brain lesions. Baló’s concentric sclerosis is a type of demyelination with pathologically alternative bands of demyelination and preserved myelin. 41. e. rarely causes a syndrome of acute flaccid paralysis. West Nile virus infection is usually asymptomatic; less than 1% develop neurological symptoms. Elderly and immunocompromised patients are at most risk for neurological complications. Imaging is nonspecific but may show changes on MRI in the basal ganglia, thalami, brainstem, and cerebellum. T2 signal change may be seen in the spinal cord. It can cause a polio-like acute lower motor neuron syndrome with flaccid paralysis. It can also present with meningitis, encephalitis, or myelitis. 42. e. is due to defective viral maturation in the brain. This syndrome is known as subacute sclerosing panencephalitis (SSPE). There is no treatment, and prognosis is progression to death over 1 to 3 years. EEG shows high-amplitude spike or slow-wave bursts correlating with myoclonus progressing to burst-suppression pattern. Slow viral infections cause conditions such as CJD, fatal familial insomnia, and Gerstmann-Straussler-Scheinker syndrome. Congenital rubella syndrome can cause mental retardation, cataracts, sensorineural hearing loss, and congenital heart disease. 43. b. Mees’ lines 44. a. Alopecia 45. d. Burtonian line 46. c. Bradykinesia 43–46. Arsenic causes acute GI symptoms, an axonal sensory neuropathy beginning 5 to 10 days after ingestion, nail changes called Mees’ lines, and hyperkeratosis and sloughing of skin from the palms and soles. Thallium poisoning has a hallmark of alopecia and may cause cranial nerve and autonomic neuropathy. Lead poisoning may cause Burtonian lines or lead lines in the gums, and causes a chronic axonal motor neuropathy, abdominal pain, and anemia. Manganese poisoning may cause an extrapyramidal syndrome. 47. d. are seen in a variety of encephalopathies. Triphasic waves on EEG are seen in hepatic encephalopathy but may be seen in other acute encephalopathies and are evidence of diffuse brain dysfunction. CJD may cause periodic sharp waves that can sometimes be mistaken for triphasic waves. A burst-suppression pattern may be seen as a late finding in status epilepticus. Triphasic waves do not suppress with benzodiazepines, but epileptiform sharp waves may at times suppress with this treatment. 48. e. It provides smooth muscle relaxation via beta-adrenergic receptors. The hypogastric nerve provides smooth muscle relaxation via beta-adrenergic receptors. The preganglionic efferent nerves exit T10-L2. Ganglia are paraganglia (next to vertebrae), preganglia (between vertebrae and end organ) or peripheral ganglia (in end organ). Nerves travel within hypogastric nerve to the inferior pelvic plexus, where they modulate urethral smooth muscle contraction and inhibit parasympathetic activity. Release of norepinephrine stimulates beta-3 adrenergic receptors in the bladder, causing relaxation (storage). Release of norepinephrine stimulates alpha-1 adrenergic receptors in the involuntary sphincter, causing sphincter contraction (storage). 49. e. A lesion of cervicomedullary junction Downbeat nystagmus is characteristic of a cervicomedullary lesions and is often seen in a Chiari malformation. A lesion of the dorsal midbrain can cause Parinaud’s syndrome with limited of vertical gaze and convergence retraction nystagmus. A lesion of Mollaret’s triangle may cause palatal nystagmus (Mollaret range red nucleus, inferior olive, dentate nucleus). A massive pontine lesion can cause ocular bobbing. A lesion of the parasellar region may cause seesaw nystagmus. 50. d. causes episodic vertigo. Ménière’s syndrome is caused by distension of the endolymphatic system (endolymphatic hydrops). Symptoms include fluctuating hearing loss at low frequencies, tinnitus, episodic vertigo, and a sensation of pressure in the affected ear. Bonus Answers 1. b. Laboratory findings show hypernatremia with inappropriate urine osmolality. Syndrome of inappropriate antidiuretic hormone secretion: usually a diagnosis of exclusion; hyponatremia with plasma osmolality less than 275 mOsm/kg H2O and inappropriate urine osmolality (>100 mOsm/kg H2O); with normal renal function; with euvolemia; without adrenal insufficiency, hypothyroidism, or diuretics. It is caused by CNS infections, tumors, and trauma; pulmonary and mediastinal infection and tumors; and drugs—phenothiazines, tricyclic antidepressants, desmopressin, oxytocin, salicylates, nonsteroidal anti-inflammatory drugs. Diagnosis in difficult cases can be aided by water loading test (oral water load of 20 mL per kg body weight in 15–20 minutes, inability to excrete 80%–90% of the oral load in 4–5 hours, and inability to suppress the urine osmolality to <100 mOsm/kg H2O). Treatment: 3% NaCl in acute severe symptomatic hyponatremia; restriction of free water intake if chronic, with option of vasopressin 2 receptor antagonist if not responding. 2. c. Deletion of 1p19q causes a better treatment response. Oligodendrogliomas account for 5% of intracranial gliomas. They usually occur in the cerebral hemispheres and are most frequent between 30 and 50 years of age. The pathology shows a typical fried-egg appearance, delicate vessels, and calcification. Deletion of 1p19q provides a better treatment response. 3. d. GlyR 4. b. anti-Hu 5. a. GAD-65 6. c. VGCC 3–6. A progressive encephalomyelitis can be seen in Hodgkin’s lymphoma associated with GlyR antibody. A sensory neuropathy can be seen with anti-Hu (ANNA 1) antibodies. Stiff-person syndrome is commonly associated with GAD-65 antibodies. Lambert-Eaton syndrome commonly is associated with voltage-gated calcium channel antibodies. 7. c. Approved for treatment of TD: most effective antipsychotic Clozapine (Clozaril®) is the most effective antipsychotic; NB: clozapine causes no EPS; safely treats PD-related psychosis; approved for treatment of TD; side effects: NB: idiosyncratic agranulocytosis (~1% incidence), weekly complete blood count for 6 months, then biweekly complete blood count for duration of treatment, hold or discontinue clozapine if white blood count or neutrophil count declines; NB: lowers seizure threshold (0.7%–1.0% per 100-mg daily dose), severe anti-ACh and anti-H1 side effects—sialorrhea (excessive salivation). 8. b. trinucleotide repeat at chromosomal locus Xq27.3. Fragile X syndrome is caused by trinucleotide repeat at chromosomal locus Xq27.3. Incidence: 1/1,000 males, 1/2,000 females. Diagnostic features include long head, large ears, hyperflexible joints, macroorchidism, short stature, and mild to severe intellectual disability; patients are often gregarious and pleasant; females are usually less severely affected. There is a high frequency of comorbidity with ADHD and pervasive developmental disorders. Fragile X is the most common cause of inherited intellectual disability. Nearly all affected boys manifest attention deficit disorder (ADD) and have learning disabilities. The most common neurocognitive symptoms are problems with abstract reasoning, complex problem solving, and expressive language; 33% meet criteria for autism. 9. c. retention of reactive pupils. Anton’s syndrome is due to bilateral occipital lobe lesions. It causes inability to see with absent response to visual threat. Patients are often unaware of having a deficit and may confabulate reasons for inability to identify objects. Pupillary reactions are spared, as this pathway bypasses the neocortex (Edinger-Westphal nucleus of the midbrain). Apraxia of the left hand may occur with anterior corpus callosum lesions. Right homonymous hemianopsia occurs with left retrochiasmal lesions. Prosopagnosia may occur with ventral occipitotemporal lesions. Visuospatial disorientation often occurs with parietal lesions. 10. d. is associated with hyperorality. Klüver-Bucy syndrome can be seen with bilateral temporal lobe damage, usually in the anterior temporal regions. Hyperorality, hypersexuality, apathy, hypermetamorphosis (overly sensitive to minute stimuli in the environment with preoccupation with these stimuli), and visual agnosia are common. Patients are unable to ignore visual stimuli.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


