Primary Central Nervous System Tumors
Edward J. Dropcho
More than 40,000 new cases of primary CNS tumors are diagnosed each year in the United States. Brain tumors are the second most frequent cause of cancer-related death among children. Brain tumors may affect adults at any age, and often have a devastating effect on patients and their families. This review will summarize the current clinical approaches to the most common primary brain tumors. For most if not all brain tumors the therapeutic strategies are in a state of evolution, and there are more than a few uncertainties and controversies. The past several years have seen an explosion of new knowledge of the molecular genetics and basic biology of brain tumors. Although the clinical progress seems painfully slow, these new discoveries are gradually being translated into more precise disease stratification and into targets for new therapies.
I. GLIOBLASTOMA AND ANAPLASTIC ASTROCYTOMA
A. Course of disease.
The World Health Organization (WHO) stratifies malignant astro cytic neoplasms into two main groups, anaplastic astrocytoma (AA, WHO grade 3) and glioblastoma (GBM, WHO grade 4) based on the degree of hypercellularity, nuclear pleomorphism, mitoses, microvascular proliferation, and necrosis. GBM is the most common glioma in adults, accounting for 50% to 60% of all cases. Unfortunately, this tumor is also the most deadly. AA accounts for 15% to 20% of adult gliomas. There is a slight male predominance. Peak age incidence for GBM is approximately 55 years and for AA about 45 years. Recent epidemiologic evidence suggests an increasing incidence. Approximately 10% of patients with AA or GBM had a prior histologically proven diagnosis of astrocytoma or other lower-grade glioma.
Patients with GBM or AA generally present with a fairly short history of some combination of headache, seizures, and focal neurologic symptoms determined by the tumor location. Malignant gliomas appear on MR scans as an irregular mass lesion with heterogenous or ring enhancement (Fig. 52.1). There is a predilection to extend across the corpus callosum or to spread along other major white matter pathways. T2-weighted or fluid attenuated inversion recovery images typically show abnormal signal extending in an irregular shape for considerable distance beyond the margins of contrast enhancement. In most if not all patients there are infiltrating tumor cells within and beyond the area of abnormal T2/fluid-attenuated inversion recovery (FLAIR) signal. The variable topography and distance of tumor cell infiltration are serious obstacles to attempts at surgical resection or other “focal” therapies for these tumors.
B. Therapy.
Standard treatment for patients with newly diagnosed GBM/AA is maxi mal tumor resection consistent with preservation of neurologic function, followed by limited-field radiation therapy (RT), and for most patients chemotherapy begun during or after RT. Several modern techniques facilitate the aggressive resection of gliomas and reduce the risk of neurologic morbidity for selected patients. Preoperative functional MRI, diffusion-tensor MRI, and intraoperative cortical and subcortical mapping can determine the tumor’s proximity to and involvement of motor and speech structures. Intraoperative MRI allows the surgeon to assess the degree of resection and possibly continue the resection to remove more residual tumor.
For patients with symptomatic tumor mass effect, aggressive surgery usually improves neurologic function. Whether the extent of initial resection of GBM/AA has a major impact on survival continues to be controversial. There has never been and probably never will be a prospective randomized study in which patients are randomized to undergo differing degrees of tumor resection. Most (not all) retrospective studies show a survival
advantage for patients who undergo an “aggressive” resection. The cutoff value of how much of the enhancing tumor needs to be resected to impact survival ranges from 75% to almost 100% in various studies. In studies where multivariate analysis showed the extent of resection to be an independent prognostic factor, the impact on survival was nearly always less than that for patient age, tumor histology, and pretreatment performance status.
advantage for patients who undergo an “aggressive” resection. The cutoff value of how much of the enhancing tumor needs to be resected to impact survival ranges from 75% to almost 100% in various studies. In studies where multivariate analysis showed the extent of resection to be an independent prognostic factor, the impact on survival was nearly always less than that for patient age, tumor histology, and pretreatment performance status.
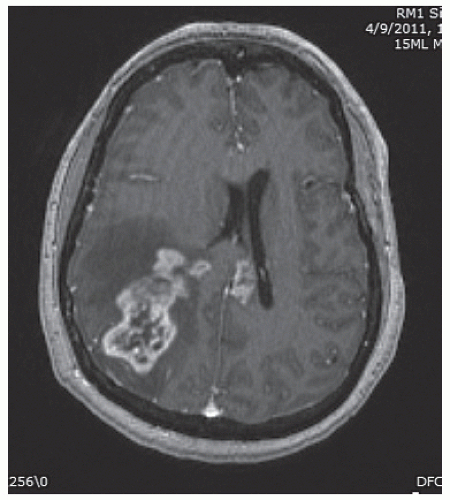 FIGURE 52.1 Axial T1-weighted MRI scan of a patient with a right parietal GBM, showing heterogeneous tumor enhancement, moderate surrounding cerebral edema and associated mass effect, and extension of tumor enhancement across the corpus callosum into the deep left hemisphere. |
Standard postoperative RT for GBM/AA is 55 to 60 Gy “focal” or “limited-field” RT delivered to a target encompassing a 2 to 3 cm margin around the radiographically visible tumor area. GBM/AA occasionally spreads through the leptomeninges or recurs far from the initial tumor site, but for the vast majority of patients the ultimate cause of death is tumor recurrence within the initial RT target area. There is no evidence that higher doses of fractionated RT or a “boost” of stereotactic radiosurgery or brachytherapy in addition to conventional RT provide any survival advantage.
Based on a randomized prospective study, the current standard chemotherapy regimen for patients with newly diagnosed GBM is daily oral temozolomide taken concurrently during RT, followed by six or more monthly cycles of temozolomide after the completion of RT. Temozolomide is an alkylating agent that has excellent oral bioavailability and shows good penetration across the blood-brain barrier. Noncumulative myelosuppression is the dose-limiting toxicity. The efficacy of this chemotherapy regimen for patients with AA has not been definitively proven, but it is a common practice to administer the same treatment as for patients with GBM. For selected GBM/AA patients, another chemotherapy option is surgical implantation of BCNU-containing wafers at the time of initial resection.
Nearly all GBMs and AAs recur despite aggressive multimodality treatment. The median time to tumor progression after initial diagnosis of GBM is 6 to 9 months. For selected patients with relatively young age, good performance status, and accessible lesions, a second surgical resection can “set up” further chemotherapy, may improve neurologic function, and modestly prolongs survival. Depending on tumor size and location, some patients may benefit from single-dose or fractionated stereotactic radiosurgery. For many if not most patients with recurrent or progressive GBM or AA, further surgery or RT are judged not to be feasible or advisable. Systemic chemotherapy or other drug treatment is then the only option available. In the United States,
the most commonly used treatment for recurrent GBM/AA is bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor. In addition to its antiangiogenic and antitumor effect, bevacizumab is a potent antivascular permeability and anticerebral edema agent.
the most commonly used treatment for recurrent GBM/AA is bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor. In addition to its antiangiogenic and antitumor effect, bevacizumab is a potent antivascular permeability and anticerebral edema agent.
There has been intense interest in a growing number of drugs that have a cytostatic or cytotoxic effect on malignant gliomas by virtue of blocking or inhibiting growth factor receptors, intracellular or extracellular signaling pathways, angiogenesis, or tumor cell migration. To date, studies of these molecularly targeted agents for newly diagnosed or recurrent GBM/AA have unfortunately yielded disappointing results. Except for bevacizumab, none are currently used in standard therapy.
Supportive care for patients with GBM/AA includes varying doses of dexamethasone to reduce peritumoral edema and increase neurologic function, and aggressive treatment of pain and/or depression if they occur. The concepts and strategies for treating glioma-associated seizures are the same as those for treating localization-related epilepsy in general. There is no definite evidence that any particular antiepileptic drug is differentially effective for glioma-related seizures versus epilepsy caused by other structural brain lesions. For patients taking dexamethasone or receiving chemotherapy agents metabolized by the liver, the nonenzyme-inducing antiepileptic drugs (e.g., levetiracetam or valproate) may offer fewer drug interactions than enzyme-inducing drugs (e.g., phenytoin or carbamazepine). For patients who do not have seizures at initial presentation, there is no definite evidence to support long-term prophylactic antiepileptic drugs.
C. Prognosis.
Patient age, tumor histology, and performance status are the most important prognostic factors for patients with malignant glioma. These are useful as predictors of individual patient outcome and are critically important in designing and interpreting clinical trials. With standard multimodality treatment, patients with GBM have a median survival of 12 to 18 months and only about 25% survive 24 months. Patients with AA have a median survival of 3 to 4 years.
Median survival is inversely proportional to age throughout all decades of adult life; older patients have a worse prognosis. Patients with a better performance status at the time of diagnosis have a better survival outlook than patients who present with severe neurologic impairment. Recent gene expression profiling studies have identified distinct molecular subclasses of GBM/AA, which may be associated with different survival outcome, but this is not yet part of standard clinical practice.
II. ASTROCYTOMA
A. Course of disease.
Astrocytoma, oligodendroglioma, and mixed oligoastrocytoma together comprise 25% to 30% of all gliomas in adults. These “low-grade gliomas” should not be considered “benign” tumors, as they generally lead to a fatal outcome. Low-grade (WHO grade 2) astrocytomas are poorly circumscribed and are character ized by diffuse infiltration of atypical astrocytes with hyperchromatic nuclei. Gross and microscopic boundaries are difficult to define. Most tumors contain a mixture of cells with “fibrillary,” “protoplasmic,” or “gemistocytic” morphology. Expression of the intermediate filament glial fibrillary acidic protein (GFAP), as demonstrated by immunocytochemical staining, is a marker for astroglial derivation.
The median age at the diagnosis of supratentorial astrocytoma in adults is 35 to 40 years, which is significantly younger than for AA or GBM. There is a slight male predominance. Since the advent of MR scanning in the mid-1980s, at least 70% of patients with astrocytoma present with seizures and no headache or other neurologic symptoms. Astrocytoma usually appears as a poorly demarcated mass lesion hypointense on T1-weighted MR images and hyperintense on T2-weighted and FLAIR images. Gadolinium enhancement is present in 10% to 30% of cases. Infiltration of tumor cells nearly always extends beyond the margins of radiographically visible tumor.
B. Therapy.
Few of the key issues regarding treatment for patients with low-grade astro cytoma have been studied in well-designed prospective or randomized clinical trials. It is therefore difficult to dogmatically state the “conventional” treatment for these tumors.
The proper treatment for patients needs to be individualized and based on several factors, including patient’s age, clinical presentation, tumor size and location, and tumor histology.
The proper treatment for patients needs to be individualized and based on several factors, including patient’s age, clinical presentation, tumor size and location, and tumor histology.
The fact that MRI scanning identifies patients with astrocytoma early in their natural course raises the question of whether all patients require immediate treatment when the lesion is discovered. Currently, the unequivocal indications for early intervention for patients with a presumed or proven astrocytoma include neurologic signs and symptoms other than seizures; presence of significant mass effect on neuroimaging; growth of the lesion on serial scans; and patient age ≥50 years. It is unclear whether the presence of MRI contrast enhancement should be an indication of early treatment, assuming the area of enhancement is biopsied and shown to be histologically low grade. For younger patients with astrocytoma who have no neurologic symptoms other than seizures, it is reasonable to defer surgical resection or RT until clinical or radiographic tumor progression occurs. There is no clear evidence that deferring treatment in this subset of patients has a negative impact on overall survival or on the likelihood of malignant tumor transformation.
Surgical resection or RT are the main treatment options for astrocytoma. Surgery is rarely curative. The impact of the extent of surgical resection on patients’ ultimate survival remains a matter of controversy. There has never been a prospective study in which “ideal candidates” with astrocytoma were randomly assigned to undergo varying degrees of surgical resection. Several retrospective series indicate a survival advantage for patients who underwent extensive surgical resection as compared with those who had only biopsy or “partial” tumor excision. In other series, the extent of surgery was not an independent predictor of survival. It is difficult to interpret these retrospective series because of a considerable selection bias: patients who have extensive resection are more likely to be younger, have better performance status, and have small, unilateral, relatively well-circumscribed tumors in noncritical locations than patients who have more conservative surgery.
There is also uncertainty whether all patients with astrocytoma should receive RT early in their course. In a large prospective multicenter study by the European Organization for Research and Treatment of Cancer (EORTC), patients with newly diagnosed astrocytoma (or oligodendroglioma) were randomized to either receive 54 Gy RT immediately after initial biopsy or resection, or to receive no RT until tumor progression. There was no difference in overall survival of patients who received early RT compared with patients in whom RT was deferred. There is no definite evidence that early RT benefits the subset of astrocytoma patients who undergo only biopsy versus surgical resection. Subsequent analysis of the EORTC study data showed a statistically significant prolongation in the time from initial diagnosis to tumor progression among patients who received early RT. These findings have been criticized for methodologic shortcomings because the study was not adequately designed to assess time to tumor progression as an endpoint.
If early RT for astrocytoma does not prolong survival but actually does delay tumor progression, it might be argued that early RT would delay tumor-related decline in patients’ neurologic function. The counter argument is that RT itself can cause neurotoxicity and should therefore be delayed as long as possible. Long-term neurocognitive toxicity of RT is a significant concern for patients with low-grade gliomas, because at the time of initial diagnosis most patients are young, have mild or no neurologic deficits, and have an anticipated survival of at least several years. The few published studies of serial neuropsychological testing of “long-term” survivors show conflicting results as to whether RT causes significant neurocognitive deficits. Recent evidence suggests worsening neurocognitive function as patients are followed for many years after RT. The question of whether early RT is more likely to have a positive or a negative effect on patients’ long-term neurologic function and quality of life is still unanswered.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


