CHAPTER 95 PRION DISEASES
Although the human forms of prion disease, including Creutzfeldt-Jakob disease (CJD), are rare, this group of fatal disorders of the central nervous system has become the subject of extensive scientific and public interest. Prion diseases affect both humans and animals (Table 95-1); are caused by infectious agents, which may consist only of a host-encoded protein; and share properties that pose a major challenge to animal and public health (Table 95-2). The critical difference between prion diseases and other neurodegenerative conditions is that they are transmissible experimentally and, sometimes, naturally. The hypothesis that bovine spongiform encephalopathy (BSE) has been transmitted from cattle to the human population as variant CJD (vCJD)1 and the possibility of secondary transmission through blood transfusion2,3 have increased the level of interest in and concern about these diseases.
| Human | Animal |
|---|---|
| Sporadic | Infectious |
| Sporadic CJD | Scrapie in sheep and goats |
| Genetic | Transmissible mink encephalopathy |
| Genetic CJD | BSE |
| Gerstmann-Sträussler-Scheinker disease | |
| Fatal familial insomnia | |
| Infectious | |
| Kuru | |
| Iatrogenic CJD | |
| Variant CJD* |
BSE, bovine spongiform encephalopathy; CJD, Creutzfeldt-Jakob disease.
TABLE 95-2 Characteristics of Prion Diseases
ETIOLOGY AND PATHOGENESIS
According to the prion hypothesis, PrPSc is the infectious agent and acts as a template for its own replication without additional informational material such as DNA or RNA.4 Evidence of the synthetic production of infectious PrP provides strong support to this hypothesis,5 although whether this theory adequately explains the existence of multiple strains of infectious agent remains controversial. After exposure to infection, there is a prolonged incubation period, extending to years or even decades in natural disease, during which there is no evidence of clinical disease, despite the accumulation of PrPSc and infection in peripheral tissues, mainly in the lymphoreticular system. There is no host immune response and no available serological test to identify the presence of infection; this implies that infected animals may enter the human food chain and infected humans may potentially act as blood donors.
Prion diseases are unique in that they occur in sporadic, genetic, and infectious forms.4 According to the prion theory, this is explained by spontaneous conversion of the normal protein to the abnormal form in sporadic disease, by instability in protein structure in cases associated with mutations of the prion protein gene (PRNP), and by transmission of self-replicating PrPSc in infectious forms. The absence of detectable PrPSc in some types of experimental prion disease has led to the hypothesis that it is intermediate forms of PrP, rather than the end-stage protein (PrPSc or PrP27–30), identified in postmortem brain samples, that are infectious. Whatever the structure of infectious PrP, these agents are remarkably resistant to normal sterilizing techniques,6 which adds to the concerns about onward transmission of infection.
HUMAN PRION DISEASES
Epidemiology
Sporadic Creutzfeldt-Jakob Disease
This rare disease occurs worldwide with mortality rates of 1 to 2 cases per million population per year in systematic surveys.7 The cause of sporadic CJD (sCJD) is unknown, and there is no good evidence of clustering of cases to indicate an environmental source of infection and no consistent risk factors identified in case-control studies.8 sCJD occurs predominantly in the older age groups, with a mean age at death of about 66 years, but younger patients can be affected, and a handful of teenagers with sCJD have been described.
Genetic Human Prion Diseases
Genetic Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker disease, and fatal familial insomnia are all associated with, and perhaps caused by, mutations in PRNP, with variations in clinical and pathological phenotypes that are related to the specific mutation.9 Genetic human prion diseases have been found in most countries, but the frequency of these cases varies; some countries such as Slovakia, Israel, and Italy have high localized incidences.10 Overall, in genetic cases, age at death is approximately 10 years younger than that in sCJD.
Iatrogenic Creutzfeldt-Jakob Disease
This form of CJD is caused by the transmission of infection from person to person in the course of medical or surgical treatment.11 Numerically, the most frequent means of transmission have been through human dura mater grafts (168 cases worldwide) and human pituitary growth hormone (180 cases), although, in rare cases, other vectors have been implicated, including neurosurgical instruments, corneal grafts, and human pituitary gonadotrophin. Incubation periods range from 1.5 to more than 30 years and vary according to the route of inoculation.
Kuru
This human prion disease occurred in the Fore region of Papua New Guinea and was caused by transmission of infection through ritual cannibalism.12 Kuru has now largely disappearedsince the cessation of cannibalism in the late 1950s, although there are still some new cases, with incubation periods exceeding 40 years.
Variant Creutzfeldt-Jakob Disease
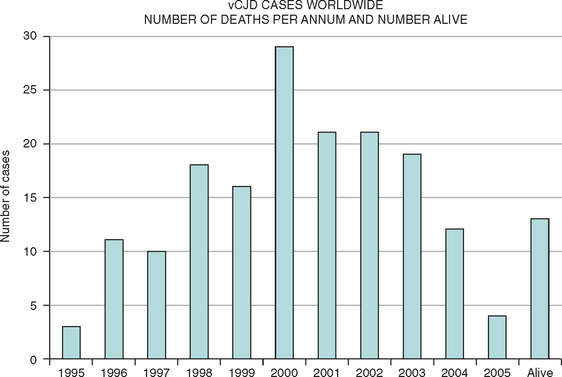
This new disease was identified in 1996, and there is now compelling evidence in support of the hypothesis that vCJD is caused by transmission of BSE to the human population, probably via the human food chain. There have been more than 150 cases of vCJD in the United Kingdom, but the annual mortality rate has dropped since 2000,13 and concerns about a massive epidemic have lessened. Small numbers of cases of vCJD have also been identified in other countries and may be related to export of bovine material from the United Kingdom, travel to the United Kingdom, or indigenous BSE (Fig. 95-1). The mean age at death in vCJD is only 30 years (range, 14 to 74 years). Possible explanations for the young age of patients with vCJD include age-related patterns of dietary exposure, age-related susceptibility, or a combination of these factors.
< div class='tao-gold-member'>






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


