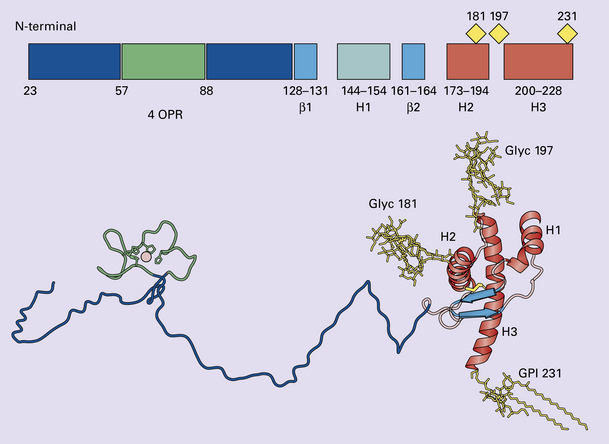
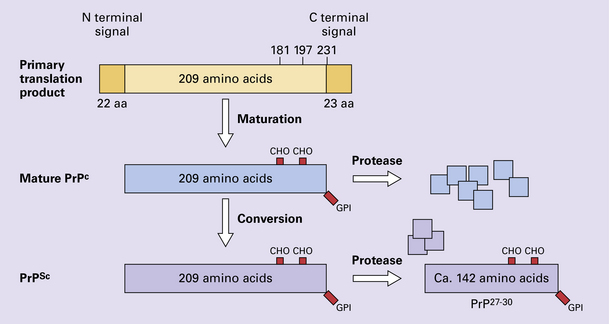
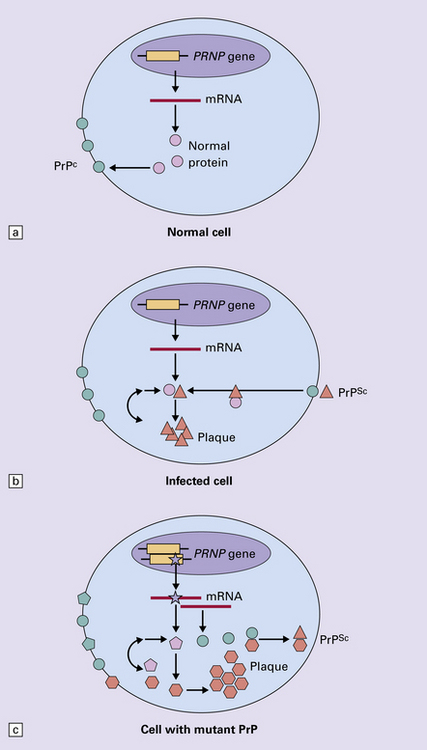
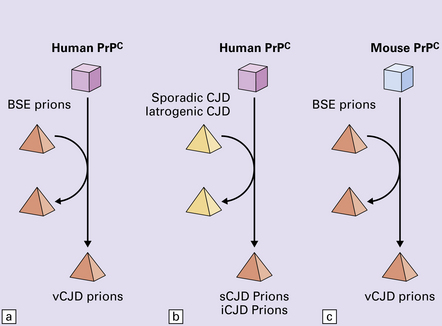
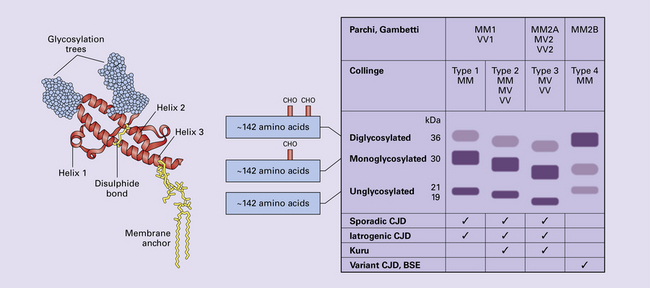
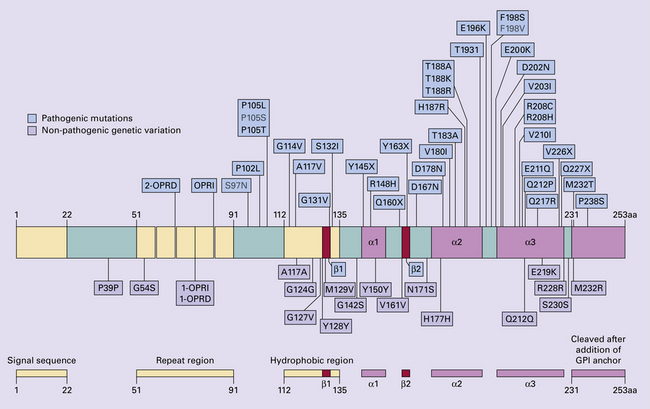
32 Common to all of these diseases are: The prion diseases have a common molecular pathology that involves the conversion of a normal cellular protein called prion protein (PrP) into an abnormal isoform (Fig. 32.1). Most evidence implicates the abnormal protein as the transmissible factor; evidence is lacking for the involvement of DNA or RNA in the infective process. 32.1 N-terminal domain: contains a large unstructured region (blue) and the octapeptide repeat domain (green), containing a copper binding site (tan-colored sphere representing the copper ion). The structured region of PrP encompasses approximately residues 124–231 and contains: (1) three α-helices (red: residues 144–154, 173–194, and 200–228); (2) a small two-strand antiparallel β-sheet (light blue: residues 128–131 and 161–164); (3) three unstructured residues at the carboxyl terminus. Glycosylation trees (yellow diamonds) are attached to residues 181 and 197 (there are un-, mono-and di-glycosylated forms.) A GPI anchor is attached to residue 231 (yellow, thick outline). The naturally occurring form of the mature PRNP gene product. Its presence in a given cell type is necessary, but not sufficient, for replication of the prion. The terms PrPC (cellular PrP) and PrPsen (proteinase-sensitive) are both used (Fig. 32.2). 32.2 PrPC and its isoforms. The primary translation product contains amino (N) terminal and carboxyl (C) terminal signal sequences, which are cleaved off during maturation. Amino acid residues 181 and/or 197 may undergo glycosylation, with the attachment of glycosylation trees (CHO, also known as glycans) at either, both or neither of these residues (i.e. resulting in PrPC which is monoglycosylated, diglycosylated or unglycosylated), and a glycosyl phosphoinositol (GPI) anchor is added to attach the PrP molecule to the cell surface. PrPSc (other terms PrP res or PrPCJD) ‘Abnormal’ form of the mature Prnp gene product (Fig. 32.3). Partly resistant to digestion by proteinase K. Believed to differ from PrPC conformationally. Often considered to be the transmissible agent or prion. The designations PrPSc (from scrapie, the spongiform encephalopathy of sheep); PrPCJD (CJD-associated PrP); PrPres (protease resistant); PrP*, and PrPP have all been used for this form of PrP. 32.3 (a,b) Conversion of normal to abnormal PrP. Endogenous or exogenous abnormal PrP can interact with normal PrP and induce it to adopt an abnormal pathogenic conformation. The abnormal form accumulates within cells, interacting with and converting more of the normal cell protein. In this way, normal PrPC is converted to PrPSc. The circles symbolize wild type PrP (mauve for immature, and blue for processed PrP), while the red triangles represent PrPSc molecules. (c) Pathogenesis of inherited prion diseases. Cells contain both mutant and non-mutant (wild type) PrP, encoded by mutant (?) and wild type alleles of the PRNP gene. Mutant PrP has a reduced threshold for conversion into pathogenic PrP. In this diagram, the blue circles represent processed wild type PrP, the mauve pentagons mutant, non-pathogenic PrP molecules, and the red hexagons mutant PrP molecules that have adopted a pathogenic conformation. The mutant, pathogenic PrP (red hexagons) is capable of converting non-mutant, non-pathogenic PrPC (blue circles) into non-mutant, pathogenic PrPSc (red triangles). PrP in normal function and disease 32.4 Mechanisms believed to give rise to PrPSc. (1) According to the template-directed refolding hypothesis, exogenously added PrPSc serves as a template and lowers the energy barrier for the conversion of PrPC into more PrPSc. In contrast, a high activation-energy barrier prevents spontaneous conversion at detectable rates. (2) The seeding (or nucleated crystallization) model hypothesizes that the conversion is reversible. However, PrPSc is stabilized by the formation of a crystal-like ‘infectious’ seed, and once this has formed, there is rapid accumulation and stabilization of further monomers. Distinct prion strains can be identified with characteristic patterns of CNS pathology and distinct incubation times. Such strains can be stably propagated in experimental animals that are homozygous for their PrP genes (Fig. 32.5). 32.5 Strains in prion diseases: a characteristic, but still unexplained feature of these diseases. The principle of strain preservation applies to transmission within one species as well as across species. (a) Transmission of BSE prions into humans has caused vCJD. vCJD prions have identical properties to those of BSE. (b) Human prions transmitted into humans generate prions that are indistinguishable from each other. Characteristically however, they generate a type 3 banding pattern and produce characteristic histological features – small or medium sized plaques and perineuronal networks located in deep cortical layers. (c) Transmission of BSE prions into mice produces mouse BSE prions that are identical to vCJD prions. Differences in the pattern of PrP glycosylation Research has revealed variations in the pattern of PrP glycosylation in CJD and in spongiform encephalopathies in animals. Western blot analysis of PrP from affected brain tissue shows three bands, corresponding to protein with two, one, or no attached polysaccharide chains (Fig. 32.6). 32.6 Glycoforms of PrP and electrophoretic patterns of prion protein in western blots. Left: PrP has two glycosylation sites and can exist in diglycosylated, monoglycosylated and unglycosylated forms. During the conversion to PrPSc, the glycans (glycosylation trees) are preserved. Right: To detect PrPSc in a Western blot, the tissue homogenate (which contains both PrPC and PrPSc) needs to be treated with proteinase K to digest the protease-sensitive PrPC but retain the partially protease resistant PrPSc, leaving a fragment of approximately 142 amino acids. Separation of the ~142-AA fragment by western blotting reveals three bands. The types of PrPSc can be further characterized according to the amino acid – methionine (M) or valine (V) – that is encoded at position 129 in PrP by each of the patient’s two PRPN alleles. The combination of electrophoretic mobility and codon 129 genotyping allows discrimination between different types human of CJD: types 1, 2, 3 or MM1/VV1 and MM2/MV2/VV2 are seen in sporadic CJD, Kuru and iatrogenic CJD, while a distinct, highly characteristic diglycosylation-dominant banding pattern is seen in vCJD, in BSE and other forms where BSE prions were transmitted accidentally or experimentally (type 4 or type MM2B). This diglycosylation-dominant banding pattern is regarded as strong evidence that BSE is the origin of vCJD. Variable glycotype patterns can be found in a single patient. A large, detailed study of 4200 samples from 200 brains showed that two types of PrP coexist in about 35% of sCJD cases. PrPSc types 1 and 2 co-occur more frequently in the MM than in the MV or VV genotypes. These molecular findings correlate to some extent to the histological phenotype. (Adapted from Parchi P, Strammiello R, Notari S, et al. Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathol 2009; 118:659–671) The commonest phenotype of prion disease is CJD. Patients typically present with subtle motor signs, which herald severe cerebellar ataxia, and progress to global dementia in under 1 year. Criteria for the clinical diagnosis of CJD have been proposed and widely adopted (Table 32.1). Table 32.1 Clinical criteria for the diagnosis of CJD aDepression, anxiety, apathy, withdrawal, delusions. bThis includes both frank pain and/or unpleasant dysesthesia. cGeneralized triphasic periodic complexes at approximately one per second. dSpongiform change and extensive PrP deposition with florid plaques, throughout the cerebrum and cerebellum. Several phenotypes of prion disease other than CJD have been identified (Table 32.2). In all of these, the mainstay of diagnosis is clinical examination supplemented by additional radiologic, electrophysiologic and neuropathologic investigations (Table 32.3). Several point mutations and insertions have been identified in the PrP gene that increase the susceptibility of PrP to assume a pathologic conformation (Fig. 32.7). 32.7 Schematic diagram of PRNP. The domains and tertiary protein structures are shown in the lower part of the figure. The upper part indicates pathogenic mutations (red) and non-pathogenic genetic polymorphisms (green). The gray text indicates PRNP variations found in cases of neurodegenerative disease in which the diagnosis was not prion disease (From Beck JA, Poulter M, Campbell TA, et al. PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 2010; 31(7):E1551–E1563) Nomenclature of PrP gene mutations This takes the form of disease phenotype (original amino acid, codon position, substituted amino acid), for example GSS (P102L). Polymorphism at codon 129 acting as a susceptibility factor In addition to these pathogenic mutations, a polymorphism at codon 129 (which codes for either valine or methionine) acts as a susceptibility factor, modulating the facility with which PrPC assumes an abnormal conformation when interacting with exogenous abnormal PrPP (see below). The frequency of this polymorphism in Caucasian populations is: In the Japanese population the frequency is: The brain may appear macroscopically normal, even in cases with long clinical histories. Most cases, however, show some atrophy (Fig. 32.8) and this may be severe, with a reduction in brain weight to as low as 850 g. In such cases, ventricular enlargement is marked and the atrophy often includes the caudate nucleus and thalamus. The hippocampus may be relatively spared, even in cases with severe atrophy elsewhere. 32.8 CJD. (a) The brain may appear macroscopically normal, but there is usually some atrophy, as here. (b) Rarely, atrophy is marked. The histologic features of prion diseases are:
Prion diseases
 Creutzfeldt–Jakob disease (CJD) and variant Creutzfeldt–Jakob disease (vCJD).
Creutzfeldt–Jakob disease (CJD) and variant Creutzfeldt–Jakob disease (vCJD).
 Gerstmann–Sträussler–Scheinker disease (GSS).
Gerstmann–Sträussler–Scheinker disease (GSS).
 Fatal familial insomnia (FFI).
Fatal familial insomnia (FFI).
 The accumulation of an abnormal form of a cellular protein (prion protein).
The accumulation of an abnormal form of a cellular protein (prion protein).
 Neuronal death, synaptic loss, and microvacuolation (spongiform change) in the brain.
Neuronal death, synaptic loss, and microvacuolation (spongiform change) in the brain.
 The accumulated PrPSc is abnormal in that it is relatively resistant to degradation in vitro by proteinase K – this property is the basis of detection of abnormal PrP by immunohistochemical techniques.
The accumulated PrPSc is abnormal in that it is relatively resistant to degradation in vitro by proteinase K – this property is the basis of detection of abnormal PrP by immunohistochemical techniques.
 According to the template-directed refolding hypothesis (Fig. 32.4), exogenously added PrPSc serves as a template for the conversion of PrPC into more PrPSc. This conformational change is kinetically controlled in that a high activation-energy barrier prevents spontaneous conversion at detectable rates.
According to the template-directed refolding hypothesis (Fig. 32.4), exogenously added PrPSc serves as a template for the conversion of PrPC into more PrPSc. This conformational change is kinetically controlled in that a high activation-energy barrier prevents spontaneous conversion at detectable rates.
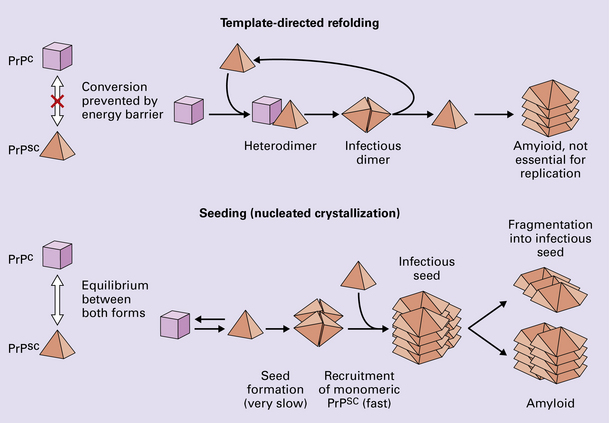
 The seeding (or nucleated crystallization) model (Fig. 32.4) hypothesizes that the conversion is reversible. PrPSc stabilizes when it form a crystal-like seed. Once a seed is formed, further monomers add rapidly.
The seeding (or nucleated crystallization) model (Fig. 32.4) hypothesizes that the conversion is reversible. PrPSc stabilizes when it form a crystal-like seed. Once a seed is formed, further monomers add rapidly.

 PRP GENE AND PATHOGENESIS OF PRION DISEASE
PRP GENE AND PATHOGENESIS OF PRION DISEASE
 CJD occurs throughout the world and has an annual incidence of 1–2 cases/million population.
CJD occurs throughout the world and has an annual incidence of 1–2 cases/million population.
 Approximately equal numbers of men and women are affected.
Approximately equal numbers of men and women are affected.
 About 10% of cases are familial.
About 10% of cases are familial.
 The age of onset is usually 55–75 years with a peak incidence in the 7th decade.
The age of onset is usually 55–75 years with a peak incidence in the 7th decade.
 Rare cases with an onset as young as 16 years or as old as 85 years have been reported.
Rare cases with an onset as young as 16 years or as old as 85 years have been reported.
PATHOLOGY
MACROSCOPIC APPEARANCES
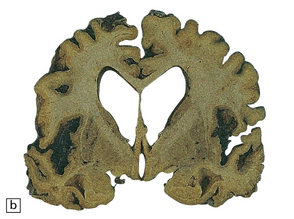
MICROSCOPIC APPEARANCES
 Spongiform change (intraneuronal vacuolation).
Spongiform change (intraneuronal vacuolation).
 Loss of synapses and subsequently of neurons.
Loss of synapses and subsequently of neurons.
 Paucity or absence of lymphocytic inflammation.
Paucity or absence of lymphocytic inflammation.
 Hyperphosphorylation of tau (immunohistochemical detection only).
Hyperphosphorylation of tau (immunohistochemical detection only).
 Accumulation of abnormal PrP (PrPSc) in various patterns:
Accumulation of abnormal PrP (PrPSc) in various patterns:
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Prion diseases


 CELL BIOLOGY OF PRION DISEASE
CELL BIOLOGY OF PRION DISEASE



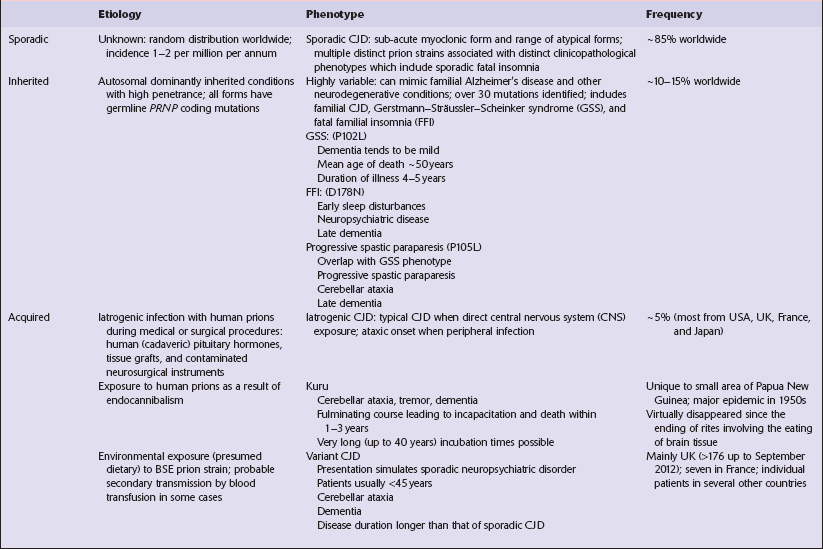







 EPIDEMIOLOGY OF CJD
EPIDEMIOLOGY OF CJD



Only gold members can continue reading. Log In or Register to continue

