(1)
Asheville, NC, USA
Recall from Chap. 1 that it took a visit to Papua New Guinea in the mid-1950s. At that time a rare but fatal disease, kuru, had broken out among the primitive Fore people. In response to what was clearly an unusual happening, the epidemiologist Carleton Gajdusek (1923–2008) traveled to the region at the invitation of the local medical officer, Vincent Zigas. Once he was in the region Gajdusek discovered that the disease was being transmitted by means of funerary cannibalistic feasts involving ingestion of brain tissue.
In the meantime, in the UK and on the Continent, scrapie outbreaks affecting sheep and goats had been occurring with some regularity. This particular illness has been known for at least 250 years, and was given the descriptive name “scrapie” because of the propensity of affected animals to rub against fences in order to remain upright. It been studied with varying results and with no clear idea as to its nature except it was observed to have a genetic component. Early efforts to establish that it was transmissible failed due to the use of too-short incubation periods. A major breakthrough occurred in the late 1930s when Cuille and Chelle finally established its transmissibility using inoculations of brain and spinal cord tissue and utilizing incubation periods of up 2 years. They reported their results in a series of publications that appeared from 1936 to 1939, and hypothesized that a virus might be the causative agent.
Those finding took on added significance in 1959 when William Hadlow noted the similarity between scrapie and kuru. Those observations led to a landmark experiment in 1966 by Gadjusek in which kuru-infected brain extracts taken from humans were shown to induce the disease in chimpanzees. Similarities of these diseases to another even more obscure disease, Creutzfeld–Jakob disease (CJD), were noted and from these studies there arose a new concept—that of a transmissible dementia—a remarkable discovery for which the Nobel Prize in Physiology and Medicine was awarded to Gajdusek and again later to Stanley Prusiner.
So what is the causative agent of the transmissible spongiform encephalopathies (TSEs) as they are known? That question was addressed by Alper in a series of experiments in which he subjected the isolated scrapie agent to ionizing radiation and UV in order to estimate its size. The result was another surprise—the agent appeared to be too small to be a virus or to involve a nucleic acid. This left proteins acting without any nucleic acid carrier of genetic information as the most likely agent. That study led to a theoretical investigation in 1967 by Griffith who proposed three possible scenarios in which a conformational change from a normal protein to a disease-causing form might take place. That is, he postulated that the disease might be caused by a self-replicating protein. Given the striking nature of that concept it is perhaps not surprising that it was simply rejected by the vast majority of the researchers in the field.
Progress on resolving the conundrum of the causal agent(s) was made by Stanley Prusiner with the development of the means of preparing greatly purified samples of the scrapie agent. Analysis of the resulting material further reduced the potential molecular mass of the molecules and made it less likely that any genetic material was present in the samples. The resulting proteinaceous, rod-like material was given the name “prion” in 1982 by Prusiner. As explained by him, that term stands proteinaceous infectious particle, symbolized as PrP. The scrapie-causing particles, designated as PrPSc, were shown to be resistant to inactivation by procedures that modify nucleic acids further reducing the possibility that a nucleic acid was involved. That effort was followed a few years later in 1985 with the cloning of the gene that encoded a 33–35 kDa, soluble and protease-sensitive protein, PrPC derived from normal and scrapie-infected brains. That effort was accompanied by yet another study that identified a 27–30 kDa protease-resistant core (amino acid residues 90–231) belonging to the 33–35 kDa disease-causing PrPSc protein. A year later the genes responsible for the normal PrPC and the disease-causing PrPSc were shown to be the same.
The goal of this chapter is to explore what has been discovered about this remarkable protein, the diseases it causes, and the revisions in thinking about the fundamental biophysical and biochemical capabilities of proteins. Although it is expressed in multiple tissues in the body, the focus is on its behavior in the brain. The chapter begins with a quick overview of how prions reach the brain upon injection in foodstuffs. That introduction is followed by a review of the panoply of human prion disease s and followed by one of the most remarkable features that characterize the prions—the existence of prions strains encoded not in differences in primary sequence but rather in differences in protein conformation. That phenomenon is still under active investigation with several ideas advanced as to how this can actually occur and how strain differences relate to the various human prion diseases.
The central biophysical feature of the prions diseases is the conversion of the normal, or cellular, form of PrPC to the misfolded, beta-sheet enriched PrPSc through contact with the latter. It is by now well accepted that the disease-causing form of the protein can replicate itself and, consequently, act as an infectious agent. This process is a robust one that survives transit through the body and from one host to another. The precise mechanism underlying this remarkable conversion, and the possible need for cofactors , is still being explored.
An additional dimension to the prion world appeared in 1994. In that year, Wickner reported the discovery of two fungal prion s —[URE3] and [PSI+]—that were altered forms of the Ure2p and Sup35p proteins, respectively. Shortly thereafter a third fungal prion—[Het-s]—was discovered. Rather than causing a disease the fungal prions seem to function as non-chromosomal (epigenetic) units of inheritance. These prions have been of great utility to the research community and many of the insights into prion phenomena have been derived from their study. These too will be examined. This short chapter then concludes with a large thought—cell-to-cell transmission of a disorder by self-templating misfolded proteins may not be limited to PrP; it may well extend to other causal agents of systemic amyloidose s and neurodegenerative disorders.
7.1 Prions Take an Invasion Route from the Gut to the Brain
Full length, normal PrP is a cell-surface glycoprotein attached to the extracytoplasmic surface of neurons and other cell types by means of a C-terminal glycosylphosphatidylinositol (GPI) anchor. One of the first questions that may be asked is how do disease-causing forms of this protein get to the brain from the stomach? This type of passage occurs following ingestion of contaminated brain tissue in the case of kuru and contaminated cattle feed in the case of bovine spongiform encephalopathy (BSE), or mad cow disease, in the recent outbreak in Great Britain. As illustrated in Fig. 7.1 several different kinds of immune cells facilitate the uptake from the intestinal lumen. That initial step is followed by entry into the lymphatic system and from there into the nervous system and on to the brain.
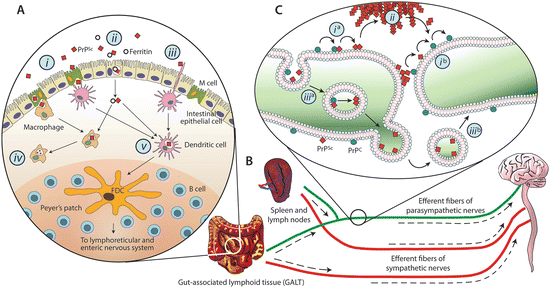
Fig. 7.1
Schematic depictions of the possible invasion routes leading from the gut to the brain taken by prions. (a) Prion uptake in the intestinal lumen by various means including: M-cell transcytosis (i), intestinal epithelial cell transcytosis (ii), or direct capture by dendritic cells (iii). In addition, while phagocytic cells such as macrophages may degrade PrPSc (iv) dendritic cells may deliver them to follicular dendritic cells where early accumulation takes place (v). (b) Amplification in the lymphoid tissues is followed by invasion of the nervous system via peripheral nerves; (c) PrPSc retrograde transport and propagation along neuronal processes either along the cell surface (i), through extracellular deposits (ii), or by means of vesicle-mediated mechanisms (iii) (from Cobb Biochemistry 48: 2574 © 2009 American Chemical Society and reprinted with their permission)
Accumulation in the lymphoid tissue (e.g., spleen, lymph nodes, tonsils, appendix, Peyer’s patches) is crucial. Several routes from the periphery to the brain are possible all involving passage from lymphoid tissue by means of the efferent fibers of the parasympathetic nerves (e.g., the vagus nerve) or sympathetic nerves (e.g., the splanchnic nerve), which regulate intestinal secretions and motility. Of particular interest are the follicular dendritic cells (FDCs), which possess extremely large surface areas. This property enables them to readily capture prion proteins. FDCs are long-lived cells, and prions can remain there for considerable periods of time, months and years.
7.2 Human Transmissible Spongiform Encephalopathy
The panoply of human prion disease s is presented in Table 7.1. One of the key features of the illnesses is the typical age of onset; another is the incubation period, and still another is the duration of the illness. The age of onset of nvCJD, 29 years, is notably shorter than that of other forms of CJD which range up to 60 years. Incubation periods also vary considerable, even within the same form of TSE. Some forms such as kuru have incubation periods that can span decades and because of that aspect prion diseases may have gone unidentified for many years if not for Gajdusek’s efforts in investigating an outbreak of an obscure illness in remote New Guinea.
Table 7.1
Transmissible spongiform encephalopathy (prion disease) of humans
Human TSE | Clinical signs | Neuropathological signs |
|---|---|---|
Sporadic forms | ||
Sporadic CJD (sCJD) | Dementia, cerebellar dysfunction | Spongiform changes, gliosis, neuronal loss, infrequent amyloid plaques |
Acquired forms | ||
New variant CJD (nvCJD) | Psychiatric symptoms | Spongiform changes in basal ganglia and thalamus, numerous amyloid plaques surrounded by vacuoles |
Kuru | Cerebellar ataxia | Spongiform changes, gliosis, neuronal loss, amyloid plaques |
Iatrogenic CJD (iCJD) | ||
– hGH associated | Gait abnormalities and ataxia | Similar to sCJD but with more numerous amyloid plaques |
– Dura associated | Resembles sCJD | Similar to sCJD |
Familial forms | ||
Familial CJD (fCJD) | ||
– Mutation and polymorphism-dependent | Personality alterations, dementia, Parkinsonism; resembles sCJD | Spongiform changes, mild gliosis, neuronal loss; similar to sCJD |
Gerstmann–Sträussler–Scheinker syndrome (GSS) | Varies with affected codon from gait abnormalities to ataxia to dementia to Parkinsonism | Numerous amyloid plaque s, neurofibrillary tangle s, gliosis, and neuronal loss |
Fatal familial insomnia (FFI) | Untreatable sleep disturbances | Profound neuronal loss and mild thalamic gliosis |
Creutzfeldt–Jakob disease (CJD) is the most common form of human prion disease with a worldwide incidence of one case per million of population per year. CJD was first described in the early 1920s. At that time Creutzfeldt described a 22-year-old female patient with dementia; his report was followed shortly thereafter by Jakob’s, which described several patients with symptoms similar to those of Creutzfeldt’s case. The PRNP prion gene is located on chromosome 20. As is the case for the other prion diseases, the specific set of clinical symptoms varies with the particular mutations, and with the presence of either methionine or valine codon at position 129. Octapeptide repeat insertions are sometimes encountered and there is another prominent polymorphism at codon 219 (glutamine or lysine) in both sCJD and fCJD. Familial forms of CJD are associated most frequently with mutations at codon 200 and less often with mutations at codons 178, 208, or 210. These mutations do not alter the native structure of PrPC as much as they do the interactions between PrPC with PrPSc and the latter’s ability to polymerize.
Gerstmann–Sträussler–Scheinker (GSS) syndrome is an autosomal dominant disease first described in 1936 by the three authors after which the disease is named. This is a heterogeneous group of disorders with a familial origin. It is most frequently associated with mutations at codon 102 and less often with mutations in several other codons of the prion gene. The codon 102 mutation leads to cerebellar dysfunction, gait abnormalities, and dementia. More generally, the specific manifestations of the illness vary depending on the particular mutation.
Fatal familial insomnia (FFI) is the least well known of the human prion disease s . It is an autosomal dominant disorder first described in 1986. It is associated with the D178N mutation of the prion gene along with presence of the methionine codon at position 129. Its most prominent symptoms are untreatable insomnia and hallucinations.
The sporadic form of CJD (sCJD) is the most common variant of TSE. It accounts for 85 % of the cases. There are two main hypotheses as to the cause of sCJD. The first is that the disease arises as a consequence of an age-dependent somatic mutation in the prion gene resulting in the formation of PrPSc. The second hypothesis is that the disease arises from a spontaneous conversion of PrPC to PrPSc in one or more neurons. These changes triggers further conversions leading to the onset of the disease. As the general term spongiform encephalopathies indicates, neuropathological indications of the disease include spongiform changes and also neuronal loss, gliosis (proliferation of glia in response to brain injury), and in some cases amyloid plaque s (Table 7.1). In many instances, the amyloid plaques are accompanied by even larger deposits of prionic aggregates that do not satisfy all the biophysical conditions for amyloids.
One of the most troubling aspects of TSE is that of iatrogenic transmission from one person to another. The first instance of this type of acquired CJD was in 1974 when a patient receiving a corneal transplant developed CJD. Other instances of iCJD involve transmission of CJD to persons through contaminated medical instruments, through the receipt of cadaveric pituitary-derived gonadotropin and human growth hormone (hGH), and to persons receiving dura matter grafts.
7.3 Animal Prion Diseases Include Scrapie and Mad Cow Disease
The classical example of an animal prion disease is scrapie, a fatal neurological disorder of sheep and goats. This disease is associated with polymorphisms at three codons in the prion protein gene—136, 154, and 171, with two combinations, VRQ and ARQ—rendering the sheep particularly susceptible to developing scrapie. As discussed earlier, this disease together with the discovery of kuru in Papua-New Guinea led to the establishment of transmissible spongiform encephalopathy as incurable neurodegenerative disorder caused by self-replicating, transmissible prions.
These diseases came to public attention in the later 1980s when two major events occurred. The first was an outbreak of a cattle prion disease, termed bovine spongiform encephalopathy (BSE), or mad cow disease, in Great Britain. By the time the epidemic ended about 5 years later 4.5 million cattle had been destroyed. The second event began in 1996/1997 with the appearance of several reports in Nature describing the emergence of not-seen-before form of CJD. Termed new variant Creutzfeldt-Jakob disease (nvCJD), this disease outbreak also took place in Great Britain. It was characterized by an earlier age of onset and with a time-course and neuropathology that differed from the classical forms of CJD. Most alarmingly, the strain responsible for nvCJD was found to be identical to that one causing bovine spongiform encephalopathy. These cases appeared followed several prominent outbreaks of BSE in the UK and elsewhere in Europe. BSE-infected meat products had apparently entered the food-chain and crossed the species barrier , thereby affecting humans. That disturbing finding along with the equally disturbing recognition of mad cow disease’s exceptionally long incubation period (up to 50 years) firmly established the disorder in the public mind. It also established that interspecies transmission can occur naturally and not just in the laboratory.
Another dimension to the prion story comes into play with studies of chronic wasting disease (CWD) of cervids—mule deer, white tailed deer, elk, and moose. Improper disposal through burial of carcasses of these animals, and also of infected cattle and sheep, have led to the introduction of prions into the natural environment. Prions released from decomposing carcasses adhere to minerals in the soil and remain bioactive for long periods of time. It is believed the improper handling of carcasses contributed to the severity of the BSE outbreak in Great Britain. In the case of mule deer, decomposing carcasses provide extra nutrients for grasses and the resulting growth attracts deer to the site. In addition to the carcass route there is a fecal-oral route of transmission. While CWD can be transmitted horizontally among cervids, it cannot be readily transmitted to humans, cattle, or sheep. That is, there is a fairly high transmission barrier in the non-cervid direction.
Transmission barrier s : In general, prions pass from one species to another far less readily than transit within the same species. This aspect gives rise to the notion of a species barrier that inhibits cross-species PrPSc transmission. The PrPC primary sequence and three-dimensional structure are both highly conserved among mammalian species. The primary impediment to cross-species transmission is thought to be a mismatch between scrapie prion and host PrPC conformations that prevent conversion of the latter to the misfolded disease-causing former. The compatibility of the two is sensitive to amino acid sequence in several critical regions so that a single point mutation in one of these regions can either promote or prevent successful seeding by the invasive prions species. Thus, primary sequence issues are a key contributor to the species barrier. In addition, glycoform properties and any other modifications may contribute to the species barrier.
Transmissible mink encephalopathy (TME) is yet another form of prion disease. This rare prion disease affects ranched mink. There are two distinct strains of TME—drowsy (DY) in which the subjects become lethargic and hyper (HY) in which they become hyperexcitable. Studies of TME carried out in the mid-1990s helped establish the strain concept, a central tenet of prion biology. Scrapie prions taken from these animals (1) exhibited distinct biochemical properties, and (2) faithfully converted normal PrPC form to new DY and HY prions.
One of the frustrations facing researchers in prion diseases is the lack of atomic level PrPSc structures. However, other pieces of information when put together point to conformational differences as being the critical factor. One of these is the variation in fragment size produced by exposure to proteinase K, a broad-spectrum serine protease that digests proteins. One of the findings from studies of TME was that DY and HY prions are cleaved by proteinase K (PK) at different amino-terminal sites. Later studied carried out using scrapie prions taken from the brains of FFI and CJD patients reinforced the emerging connections between prions, strains , conformations and PK-derived fragment lengths. In FFI, PrPSc was cleaved at residue 97 to produce 19 kDa fragments, whereas in CJD and sCJD, scrapie prions were cleaved at reside 82 to produce 21 kDa fragments. The implications from these studies were twofold—that prions did not require genetic material to convey disease, and that strains arose from differences in conformation as reflected by their varying PK cleavage sites.
7.4 There Are Multiple Prion Strains Incubation Periods and Pathology
That a single protein can give rise to multiple strains just like a conventional, nucleic acid bearing pathogen is quite remarkable, and has been the subject of considerable interest. This aspect has been studied for over 25 years in mice, in other mammalian species, and in yeasts. By the early 1990s over 20 different strains of mouse prions had been found, and strain differences were found to underlie the various forms of human TSE, as well.
Strains can be distinguished from one another by differences in disease characteristics. These include:
Clinical signs of the disease;
Differences in disease progression (e.g., incubation period, survival time);
Biochemical properties (e.g., responses to proteinase K digestion and heat);
Distribution in the brain, and
Histological features.
Differences in strains can arise in several ways. They can appear as a consequence of slight differences in primary sequence. These include tiny differences in primary amino acid sequence from individual to individual due to mutations and polymorphisms. Strains may also arise from differences in posttranslational modifications. They may arise, perhaps even more fundamentally from the intrinsic dynamics of the prion protein itself, which continually samples a host of different conformational states and substates. For example, using FRET single molecule biophysics , yeast Sup35 was found to adopt an ensemble of collapsed and rapidly fluctuating structures on a 20–300 ns timescale. This prion possesses a fluctuating amyloidogenic N-terminal region joined to an extended charged and solubilizing middle region. Putting these and similar findings together gives rise to the widely accepted hypothesis that strains are encoded in the prion’s tertiary structure, i.e., in its conformations. Importantly, strain -associated prion conformations are stabilized through formation of oligomeric seeds . They can then pass stably from one individual to another where they convert resident PrP proteins to the transmitted prion conformation, thereby giving rise to a stable phenotype.
7.5 The Protein-Only Hypothesis and Its Corollaries
The overriding understanding in the field at present is that:
1.
PrPSc is a beta-sheet rich, amyloidogenic alternate conformation of the PrP protein. In this alternative conformation, the protein is resistant to proteolysis, is detergent insoluble and, as is the situation for other misfolded disease-causing proteins, forms aggregates and amyloid-like structures.
2.
Small oligomers, either amorphous in fibrillar in organization, operate as minimal nuclei that stabilize alternative scrapie conformations.
3.
These nuclei form seeds that catalyze the conversion of normal form of the protein in the cell to the same alternative conformation.
4.
These seeds can be transmitted to other cells where they again can convert normal PrPC to an alternative PrPSc conformation.
5.
Several different alternative three-dimensional conformations can be stabilized through oligomerization. These give rise different strains , each with its distinct set of clinical manifestations and neuropathological signs.
6.
Transmission barrier s exist between individuals of the same species and between members of different mammalian species; these are determined by how well transmitted scrapie and resident normal prions overlap in primary sequence, posttranslational modifications, and tertiary structure.
Following the discovery that PrPC and PrPSc shared the same primary sequence efforts were made to firmly establish the main prion tenets, known as the protein-only hypothesis , described above. Those efforts spanning the past 20 years have largely succeeded in establishing that the above-described chain of events is the correct one. That is, prions by themselves are transmissible and infectious, and are the causal agent of the transmissible spongiform encephalopathies such as kuru in humans, scrapie in sheep, and bovine spongiform encephalopathy in cattle.
There are a number of milestones in the efforts to establish the protein-only hypothesis . One of these occurred in 1994 with a demonstration of the cell-free conversion of PrPC to protease-resistant form similar to PrPSc. The demonstration highlighted the necessary presence of PrPSc in the process, thereby providing evidence for direct PrPC–PrPSc interactions in the conversion.
A few years later, in the 2001–2005 time period, Soto and colleagues introduced a new process that greatly increased prion concentrations. In this technique called protein misfolding cyclic amplification (PMCA), a mixture containing PrPSc and a far-larger amount of PrPC is incubated to grow PrPSc aggregates. That step is followed by sonication in which the sample is exposed to ultrasound that fragments the aggregates into smaller pieces that serve as new PrPSc seeds . The two-step, grow/break process is then repeated over many cycles to produce large quantities of PrPSc aggregates. Inoculation of laboratory animals with the in vitro PMCA-derived scrapie proteins produced scrapie illness identical to that produced by brain-derived material. Additional studies focusing on prion generation and transmissibility provided further evidence for the correctness of the proposed causal chain. It was also was used a few years later to demonstrate the generation of scrapie prions from PrPC together with co-purifying lipids and synthetic polyanions without requiring the presence of PrPSc seeds. Those dramatic results served to focus attention of the role(s) of non-genetic cofactors both in cell-free systems and in the brain.
Yet another piece of data supporting the protein-only hypothesis , and linking strain effects to conformation, was provided in the late 1990s when conformation-dependent antibodies were devised. Using a conformation-dependent immunoassay, scrapie prions were first subjected to denaturation and then the fraction of antibodies that bound exposed epitopes on the prions was measured. When this technique was applied to each of eight strains of mouse scrapie, differences in binding fractions were observed, thereby providing further support for the underlying chain of reasoning.
Requirement for cofactors : As noted above cofactors may contribute to the conversion process. A variety of potential cofactors have been identified. These are primarily large biomolecules—polysaccharides, nucleic acids, and proteins. Among the more notable examples are glycosaminoglycans (GAGs) such as heparin, the nucleic acid RNA, and the membrane lipid phosphatidylethanolamine. Studies of these possible contributors to the conversion of PrPC to PrPSc have produced several tentative conclusions in spite of variations in results arising from strain and host dependencies. One of these is that most, if not all, of these cofactors are dispensable for the conversion, itself. Templated conversion can occur in their absence, but at exceptionally low rates. The cofactors interact with the PrPs; they influence their conformation and stability, and by that means accelerate the conversion.
7.6 Biogenesis of the PrP Protein
As discussed in the preceding two chapters, newly synthesized proteins are processed, subjected to quality control, and shipped to their cellular destinations. Prosthetic groups—sugars and lipids—are added to proteins destined for insertion in membranes, thereby enabling their attachment. These modifications are made subsequent to translation, in several stages. The overall process resembles an assembly line that builds up the proteins, folds them, inserts them into membranes, sorts them, labels them with targeting sequences, and ships them out.
The Golgi apparatus consists of a stacked system of membrane-enclosed sacs called cisternae. Some of the polysaccharide modifications needed to make glycoproteins are either made or started in the rough ER. They are then sent from the rough ER to the smooth ER where they are encapsulated into transport vesicles pinched off from the smooth ER. The transport vesicles are then sent to the Golgi. The Golgi apparatus takes carbohydrates and attaches then as oligosaccharide side chains to form glycoproteins and complete modifications started in the rough ER. The cellular prion protein, PrPC, is a glycosylphosphatidylinositol (GPI)-anchored protein. Like other proteins in this class, it is synthesized in the endoplasmic reticulum, processed and finished in the Golgi apparatus, and transported to the cell surface.
Most proteins destined for insertion in the plasma membrane contain covalently linked oligosaccharides that extend out from their extracellular side. These proteins are referred to as glycoproteins. There are two forms of modification, N-linked and O-linked. In N-linked glycoproteins, a carbohydrate, or glycan, is added to a side chain NH2 group of an asparagine amino acid residue. In O-linked glycoproteins, the oligosaccharide chain is appended to a side chain hydroxyl group of a serine or threonine amino acid residue. Carbohydrates are hydrophilic; addition of the N-linked glycans increases solubility of the proteins, and influences protein folding and stability. Their addition also facilitates intracellular transport and targeting, and assists in the signaling roles of the mature proteins. The fully dressed PrPC with its attached N-linked glycans at Asn-181 and Asn-197, attached copper ions, and GPI anchor is depicted in Fig. 7.2.
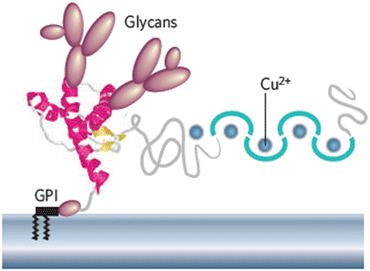
Fig. 7.2
Schematic depiction of a mouse prion protein PrP (residues 23–231). Shown are two domains—a compact C-terminal domain terminated with a GPI anchor and a natively unstructured N-terminal domain that binds copper ions. The compact domain contains three α-helices shown in red and a pair of short anti-parallel β-strands depicted in yellow (from Caughey Nature 443: 603 © 2006 Reprinted by permission from Macmillan Publishers Ltd)
7.7 Structure of the PrPC Protein
A more detailed look at the PrP’s N- and C-terminal domain structure is presented in Fig. 7.3. As can be seen in the figure, PrP contains a 22-amino-acid-residue signal peptide in its N-terminal region that is cleaved. The truncated protein containing residues 23–231 contains an N-terminal disordered region plus a C-terminal compact, globular folding core spanning amino acid residues 121–231. The folding core is predominantly alpha helical in it secondary structure and misfolds into an alternative PrPSc conformation under the influence disease-causing mutations. The four fatal prion diseases affecting humans (Table 7.1) are rare diseases brought on by missense mutations. These mutations are primarily located within the compact folding core and concentrated in the region about the H2 and H3 helices, while a few mutations causing GSS are found in the stretch from CC2 to H1. In addition, there are two polymorphisms that play a key role in the transformation from benign to lethal conformations in CJD. As remarked upon in Section 7.2, there is a codon 129 polymorphism—methionine or valine, and a codon 219 polymorphism—glutamine or lysine. These do not affect the native prion fold but when partially denatured the polymorphisms (especially the one at codon 129), strongly affect the propensity to form amyloids and the ability to propagate.
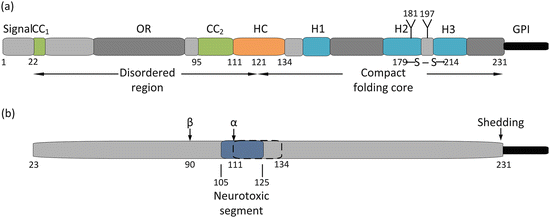
Fig. 7.3
(a) Structure of the cellular prion protein. The first 22 amino acid residues comprise a signal peptide that is cleaved leaving a mature 208 residue prion protein. Residues 23–110 constitute a disordered (random coil ) region followed by a globular, compact folding core consisting of residues 121–231 and a C-terminal GPI anchor. The N-terminal region contains a pair of positively charged acidic amino acid clusters (CC1 and CC2), and an octarepeat region (OR). The protein possesses a hydrophobic core (HC), a single disulfide bond (between residues 179 and 214), a pair of glycosylation sites (at residues 181 and 197), and three alpha helices (H1, H2, and H3). (b) Proteolytic processing of the prion protein. There are two sites where proteolytic cleavage occurs—an α-cleavage site at residues 111–112, and a β-cleavage sites in the vicinity of residue 90. In addition, the GPI anchor may be shedded to produce an anchorless prion protein, Also shown (in dark blue) is a neurotoxic segment spanning residues 105–125 that overlaps with the hydrophobic core (dashed lines)
A growing number of ligands have been identified. Some of these bind to the octapeptide repeats in N-terminal region, while others bind to specific sequences in the C-terminal core. (These are examined shortly.) The two regions are functionally coupled. They both serve in a neuroprotective capacity under normal conditions and contain segments involved in the generation of neurotoxic signals in disease scenarios.
Full-length PrP is proteolytically processed. These cleavage actions parallel those occurring in the processing of the Alzheimer’s disease-associated amyloid beta precursor protein (APP), to be studied in detail in the next chapter. Although they are not yet as well understood as the APP processing , there is accumulating evidence that these similar actions, and the fragments generated by them, have important roles in prion physiology. The reason why becomes clear in an examination of Fig. 7.3b. Two cleavage sites have been identified. The first, the α-cleavage site is located between residues 111 and 112, which lies within the segment of the protein believed to cause its neurotoxic effects. Cleavage at that location eliminates that possibility and imparts a neuroprotective role to the breakage, which generates a detached soluble N1 fragment (containing an important PrPC–PrPSc interaction site) and a remaining membrane-bound C1 fragment. Cleavage at the β-site located near the end of the octarepeat region results in the generation of a soluble N2 fragment and a membrane-bound C2 fragment containing an intact neurotoxic segment. Finally, the prion protein may undergo removal of its GPI anchor by a sheddase resulting in the release into the intracellular spaces of a largely intact prion protein.
7.8 PrP Resides in Caveolae and Lipid Rafts
Membrane lipids form gels and liquid states through the cooperative effects of multiple weak noncovalent interactions such as van der Waals forces and hydrogen bonds. As a result there is considerable fluidity of movement within the membrane, and the lipid and protein constituents are free to diffuse laterally. The overall structure is that of a fluid of lipids and membrane-associated proteins undergoing thermally driven Brownian motion. These properties were codified in the Singer–Nicolson fluid mosaic model of biological membranes introduced in 1972.
That model was challenged by biophysical studies that posited that lipids in biological membranes might exist in several phases and as a result would sequester membrane lipids and proteins in distinct microdomains . In this depiction, lateral movement may not be quite as free as presented in the Singer–Nicolson model due to mechanical constrains imposed by the cytoskeleton and through the actions of large scaffolding proteins. Instead, there is a mosaic of membrane compartments each with its own distinct population of lipids and proteins. This later model was introduced by Karnovsky and Klausner in 1982, and extended by Simons, van Meer, Ikonen, and others in the years since then.
In more detail, compartments enriched in cholesterol and sphingolipids contain high concentrations of signaling molecules—GPI anchored proteins in their exoplasmic leaflet, multiple transmembrane receptors, and numerous signaling proteins recruited to its cytoplasmic surface. The best characterized of these signaling compartments are the caveolae (little caves) and lipid raft s . The former are tiny flask-shaped invaginations in the plasma membrane. They are detergent insoluble and are enriched in coat-like materials, caveolins, which bind to cholesterol. The latter, the lipid rafts, differ from caveolae in that they are flat and do not contain caveolins. They are formally defines as small (10–200 nm), heterogeneous, highly dynamic sterol- and sphingolipid-enriched domains that compartmentalize cellular processes. These structures play an important role in signaling at synapses where they enable proteins that have to work together to be localized near one another. Prions preferentially localize to lipid rafts and to a smaller extent to caveolae. These structures and their biophysical properties are discussed further in the Appendix to this chapter.
7.9 Protective Functions of PrP
The prion protein is a synaptic protein expressed at high levels in neurons and found in both presynaptic and postsynaptic compartments. It localizes to detergent-resistant lipid raft s , and is internalized in clathrin-coated vesicles and endosomal organelles. Although its exact physiological functions are not completely characterized, a considerable body of data has been collected supporting the idea that the prion protein normally functions as a signaling platform. The protein does not penetrate to the cytoplasmic leaflet of the cell surface membrane, but is believed to partner with one or more co-receptors that initiate downstream signaling.
Under normal cellular conditions PrPC acts in a neuroprotective capacity related to its role in synaptic transmission and cell adhesion. A number of cell adhesion and extracellular matrix protein have been identified that bind PrPC. Prominent among these binding partners are heparan sulfate proteoglycans (HSPGs), vitronectin, and low-density lipoprotein receptor-related protein 1 (LRP1). These bind sequences in the N-terminal region of PrPC. Similarly, laminins and NCAM among others bind motifs in the C-terminal domain. These molecules act as neurotrophic factors that confer signals essential for growth and preservation of synapses and neurons. Binding of these ligands initiate a series of events including recruitment the tyrosine kinases Fyn or Src, which act as signaling hubs, to initiate a broad spectrum of downstream signaling actions in different neuronal populations (Fig. 7.4a).
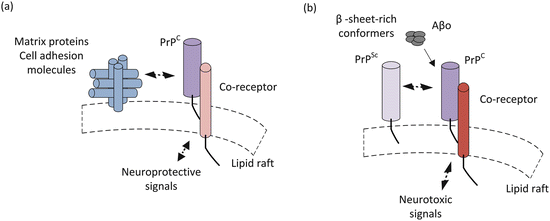
Fig. 7.4
Simplified picture of prion signaling under (a) normal and (b) disease-causing conditions
A way that cellular PrP acts in a neuroprotective capacity is through its regulation of glutamatergic synaptic transmission. One of its co-receptors is metabotropic glutamate receptor 5 (mGlu5). In response to ligand binding, PrPC-mGlu5 recruits the tyrosine kinase Fyn to the cytoplasmic face where it initiates a signaling cascade that includes regulatory phosphorylation of nearby NMDA receptor subunits. These actions protect again excessive entry of Ca2+ into neurons through the NMDA receptors. Further examples of neurotrophic actions are:
Maintenance of axonal myelination while PrP removal results in chronic demyelinating polyneuropathy (CDP),
Preservation of embryonic cell adhesion in zebrafish through its localization of E-cadherin adhesion complexes, and
Support of sensory information processing in the olfactory system.
7.10 Toxic Actions of PrP
When disease-causing mutations or other abnormal conditions are present PrPC has an increased propensity to misfold into the scrapie form PrPSc. In this alternative conformation, it interacts with the normal cellular prions to initiate abnormal signaling. This interaction seems to be a necessary one. In the absence of the cellular form, scrapie PrP does not cause neurodegeneration. As discussed earlier, PrPSc is a β-sheet-rich conformer. Interestingly, it is not the only β-sheet-rich conformer that interacts with PrPC. Another set of prominent interactors are the amyloid-β oligomers (Aβo) that have a central place in the Alzheimer’s disease pantheon. These aberrant interactions compete with the normal ones leading to shift in signaling from protective to harmful. The altered, neurotoxic signaling occurring in the presence of β-sheet-rich conformer is depicted in Fig. 7.4b. That subject is revisited in the next chapter.
Several regions within PrP have been identified as playing crucial roles in the protein’s normal neuroprotective and abnormal neurotoxic activities. The most prominent of these is the central region (CR) spanning residues 105–125. This region, highlighted in Fig. 7.3b, contains a segment enriched in charged residues followed by a sequence supplied with several hydrophobic ones. It is one of the sites bound by PrPSc and, significantly, deletion of this region renders the resulting protein, ΔCR PrP, toxic to neurons. A second PrP sequence, this one spanning residues 23–31 is also involved in binding PrPSc and its loss also enhances PrP neurotoxicity.
These results support the notion that competitive interactions promote PrPC-PrPSc toxicity. That is, the matrix proteins and cell adhesion molecules that normally bind to these regions are prevented from doing so when PrPSc binds to the same sites. Toxicity may result from failure of the blocked PrPC to convey its normal signals and/or from its redirection into a toxic signaling pathway (Fig. 7.4b).
The above mechanism is not the only contributor to prion toxicity. As is the case for most, if not all, neurological disorders, there is an accumulation of misfolded proteins, synaptic transmission fails, protein quality control declines, and neurons are lost. There is evidence that, in response to a buildup of misfolded PrP oligomers, ER stress increases, protein translation is reduced, and the UPS is inhibited. These conditions when combined with an increased sensitivity to synaptic excitotoxicity and misdirected neurotrophic signaling may be sufficient to send neurons along an irreversible apoptotic trajectory.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


