CHAPTER 24 RETINAL DISEASE
The retina reflects many of the pathophysiological processes that occur in the central nervous system (CNS). Indeed the retinal appearances may be diagnostic but, conversely, can be nonspecific; for example, pigmentary retinal changes are part of the diagnostic criteria for Kearns-Sayre syndrome but are also present in many other diseases. Furthermore, in other diseases, the retina may appear clinically normal, but psychophysical and electrophysiological testing reveal abnormalities.
Uveitis, inflammation of the uveal tract, can occur in association with neurological disease. In this chapter, the classification recommended by the Standardization of Uveitis Nomenclature Working Group is used.1 In this classification, the primary site of inflammation in anterior uveitis is the anterior chamber. For intermediate uveitis, it is the vitreous humor. The primary site of inflammation in posterior uveitis can be the retina or choroid. Panuveitis is used when there is no predominant site of inflammation, but inflammation occurs in the anterior chamber, vitreous humor, and retina and/or choroid.
UVEOMENINGEAL SYNDROMES
Sarcoidosis
Sarcoidosis is a multisystem granulomatous disorder of unknown etiology that can affect the eye and the CNS, as well as the lungs, skin, and other organs. Of all patients with sarcoidosis, 25% to 60% have ocular involvement, which may be the manifesting sign. Sarcoidosis can affect most ocular tissues, but the most common ocular manifestations are uveitis and conjunctival nodules. Uveitis can occur in about 30% of patients before nonocular signs.2
A 10-year follow-up study of patients with uveitis caused by sarcoidosis or presumed sarcoidosis revealed that over this period, the disease spread most often to the CNS.3 Posterior segment involvement is frequently associated with neurological disease.4,5 It may be more common in white patients, especially elderly women,2 although this has not been confirmed by all studies.6 The optic nerve may become swollen because of intraocular inflammation, when visual acuity is usually preserved, or it may be involved either directly by granulomas or as a result of meningeal changes, when loss of visual acuity and field result from compressive optic neuropathy.
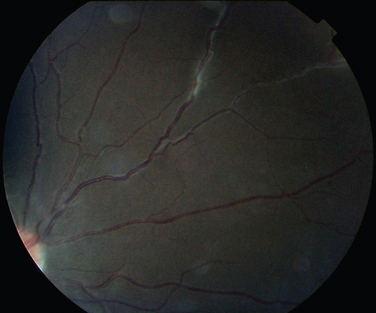
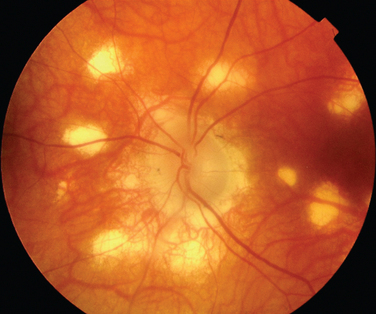
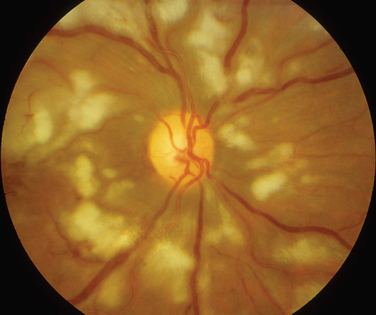
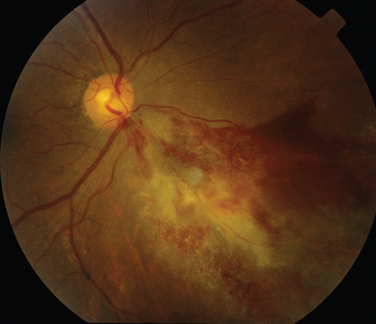
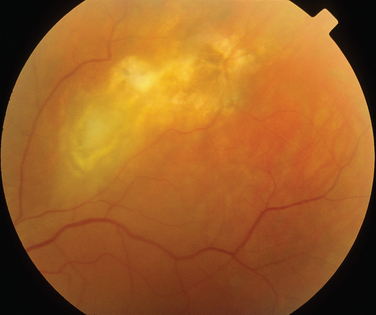
The classic sign of retinal involvement by sarcoidosis is retinal periphlebitis (Fig. 24-1),7–9 which may be described as similar to candle wax dripping. Other vascular changes include retinal hemorrhages, vein occlusions, neovascularization, and possibly retinal arteriolitis with aneurysm or ectasia formation.10 Focal subretinal lesions have been described, but these do not appear to affect vision.9 Confluent choroidal infiltrates have been described.11 Appearances similar to that of a multifocal choroiditis (Fig. 24-2),12,13 and a serpiginous choroidopathy have been described.14 Multiple sclerosis may include a similar ocular inflammatory manifestation, but the choroid is not involved.
Behçet’s Disease
Behçet’s disease is characterized by recurrent episodes of orogenital aphthae and by systemic and retinal venous thrombosis. It remains a clinical diagnosis, and diagnostic criteria have been published.15 The CNS manifestations of Behçet’s disease can be categorized into parenchymal and nonparenchymal involvement (neurovascular Behçet’s disease). Parenchymal CNS manifestations include brainstem and hemisphere involvement, spinal cord lesions, and meningoencephalitis. Nonparenchymal involvement includes dural sinus thrombosis, arterial occlusion, and arterial aneurysms. These categories have different clinical and prognostic properties.16 The ocular manifestations in the parenchymal category include optic neuritis and ischemic optic neuropathy.17–19 Papilledema caused by intracranial hypertension secondary to dural sinus thrombosis occurs in neurovascular Behçet’s disease20,21 and can result in significant visual loss. Intracranial hypertension can, however, occur in the absence of venous sinus thrombosis or meningitis.18
Inflammatory eye disease occurs in approximately 70% of all patients who may have additional neurological or neuro-ophthalmological disease. The inflammation usually occurs after the onset of oral aphthosis, but the delay between the two may be as long as 14 years.22 In approximately 10% of patients, intraocular inflammation is the manifesting feature,23 and in rare cases, neither oral nor genital ulcers may occur at all.24 Usually the involvement is bilateral.
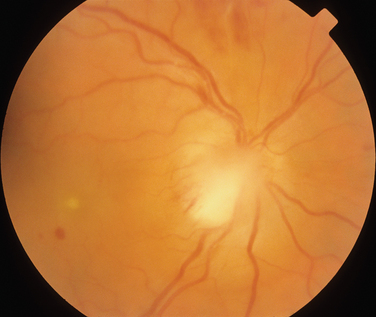
The main ocular finding is panuveitis, although there may be differing degrees of anterior and posterior segment involvement. The anterior uveitis may be so severe that the inflammatory cells precipitate in the inferior portion of the anterior chamber, forming a hypopyon. Retinal vein occlusion is the most characteristic fundal sign, but others include retinal perivasculitis and retinal infiltrates. The perivasculitis involves mainly the veins and less frequently the arteries. The retinal infiltrates are collections of lymphocytes in the superficial retina and are pathognomonic of Behçet’s disease (Fig. 24-3). They resolve spontaneously and carry no visual morbidity. However, recurrent retinal vein occlusions can result in total attenuation of all vessels and consequent optic atrophy. Macular edema may also cause visual loss. Unfortunately, the visual prognosis is poor in spite of the development of new immunosuppressive therapies. Up to 15% of patients are unresponsive to these therapies,25 and the disease may remain active for many years.
Vogt-Koyanagi-Harada Disease
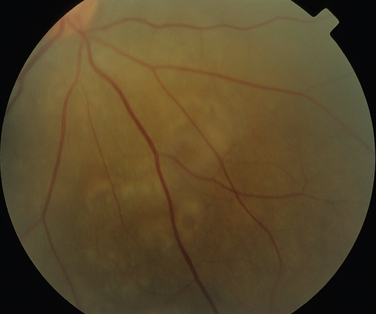
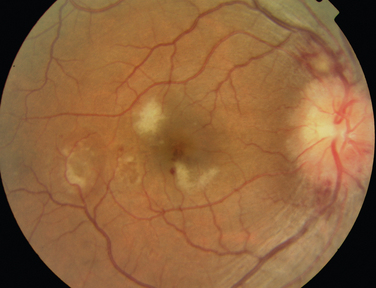
Like Behçet’s disease, it is diagnosed on clinical findings. Diagnosis can be difficult because there are no specific tests and the clinical features are dependent on the stage of the disease at which the patient is examined. Diagnostic criteria have been published.26 One of the following neurological features has to be present for the diagnosis of VKH: meningismus, tinnitus or cerebrospinal pleocytosis. The ocular features have been divided into early and late manifestations of the disease. In the early manifestation, the retinal and choroidal features are mainly in the form of serous retinal detachments. In the late manifestations, choroidal depigmentation that results in a mottled “sunset glow” appearance, nummular chorioretinal depigmented scars, and retinal pigment epithelium clumping and/or migration26 may be present (Fig. 24-4). Recurrent or chronic uveitis also occurs in the late stage of VKH. The criteria include the presence of bilateral ocular involvement. The integumentary findings are alopecia, poliosis, or vitiligo. A further neurological association with VKH is Guillain-Barré syndrome. Three patients who developed VKH within 3 months of having Guillain-Barré syndrome have been reported.27
RHEUMATOLOGICAL DISEASES AND THE SYSTEMIC VASCULITIDES
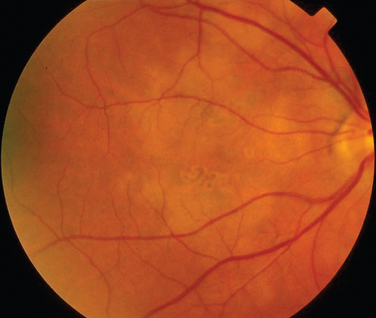
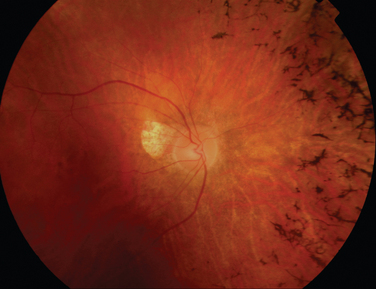
Rheumatoid arthritis is associated with corneal and sclera changes but not usually retinal ones. Retinal involvement is, however, a common ocular feature of systemic lupus erythematosus. Indeed, the presence of ocular features may mirror disease activity elsewhere.28,29 Retinal arteriolar occlusion may occur, and multiple cotton-wool spots may be seen (Fig. 24-5). In rare cases, venulitis develops. Choroidal involvement can also occur. A postmortem study of patients with systemic lupus erythematosus affecting the brain and eye demonstrated vasculitis in the brain and the choroidal vessels.30 A number of retinal arterioles were occluded with thrombus, but there was no evidence of inflammation of the retinal vessels. Emboli from a diseased heart valve or deposition of immune complexes may have caused the occlusion.
Large Artery
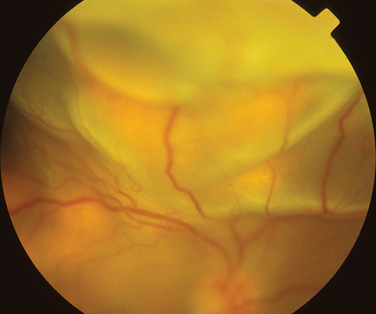
Giant cell arteritis is a necrotizing vasculitis targeting large and medium-sized arteries and affects mainly white patients older than 50 years. Anterior ischemic neuropathy is the most common cause of visual loss in giant cell arteritis, but patients can suffer from either central retinal artery occlusions or, less commonly, branch retinal artery occlusions. Compromised blood flow to the retina may result in signs of ischemia such as cotton-wool spots and hemorrhages. Posture can also affect the ischemia in that vision may be reduced in the upright position, in relation to lying down.31 Poor perfusion of the choroid may result in reduced vision, which can be reversible, but permanent infarction can occur (Fig. 24-6). Treatment is aimed primarily at preventing both visual loss in the second eye and the development of stroke.
Takayasu’s disease is another disease of the large vessels. Retinal disease is present in fewer than 50% of patients with Takayasu’s disease.32 The retinal changes include vessel dilation, tortuosity of vessels, arteriovenous anastomoses, and hemorrhages. In many patients, hypertension is probably the major cause of retinal changes; in some patients, however, the changes may result from reduced flow to the retinal circulation (i.e., a hypotensive retinopathy).32
Medium-Sized Artery
Ocular involvement occurs in 10% to 20% of patients with polyarteritis nodosa. Retinal artery occlusion, ischemic retinopathy, exudative retinal detachment,33 and choroidal infarcts have been described.34 Kawasaki’s disease tends not to affect the retina, although one postmortem study did yield evidence of retinal ischemia.35
Medium-Sized and Small Vessels
Wegener’s granulomatosis is characterized by a systemic vasculitis and necrotizing granulomatous lesions that may be found in the respiratory tract, kidney, orbit, and brain. Between 30% and 50% of patients with Wegener’s granulomatosis have neurological involvement.36,37 The patterns of neurological involvement have been described as granulomatosis invasion of the orbit, granulomas of the brain and meninges, and vasculitis of the nervous system.36 The orbital involvement usually is from the extension of disease in the paranasal sinuses. Orbital involvement may be the initial sign of the disease, manifesting with orbital pain, rapidly progressing proptosis, and limited eye movements with associated granulomatosis infiltration of adjacent structures. Cranial nerves may also be involved; the optic nerve, the abducens nerve, and the facial nerve are the most commonly affected.37 Retinal and choroidal involvement is rare but may manifest as retinitis, vein occlusion, or an intermediate uveitis.38 Some of these changes, however, may have been secondary to hypertension or cytomegalovirus–associated retinitis. A number of patients taking cyclophosphamide for Wegener’s granulomatosis have developed cytomegalovirus-associated retinitis (Fig. 24-7).39–41 In patients with the limited form of Wegener’s granulomatosis, the neuro-opthalmological features may, again, be the earliest feature of the disease.42 A similar pattern of neuro-ophthalmological involvement is seen, the orbit being affected more frequently than the retina.43
Ocular involvement in the Churg-Strauss syndrome can be in the form of either chronic orbital pseudotumor or ischemic vasculitis. The retinal manifestations of the latter group include retinal artery occlusions.44 Cotton-wool spots in the retina have been described in a patient with microscopic polyangiitis. The patient also had a very severe uveitis that resulted in a hypopyon.45
MULTIPLE SCLEROSIS
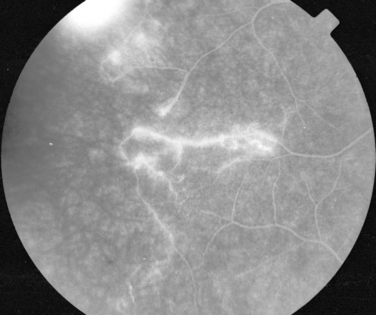
Manifestations of multiple sclerosis in the eye other than optic neuritis include uveitis and retinal changes, such as periphlebitis or sheathing or cuffing of the retinal veins by lymphocytes and plasma cells.46–49 This may occur in isolation or as part of an intermediate uveitis. Periphlebitis may occur in up to 36% of patients with multiple sclerosis50,51 and is usually asymptomatic. These changes are often in the peripheral retina and so cannot be viewed through a direct ophthalmoscope trained on an undilated pupil. Indeed, the changes may be visible only on fluorescein angiography (Fig. 24-8).52 Focal perivenous hemorrhage can also occur. The presence of vascular changes and/or ocular inflammation in patients with optic neuritis is associated with a greater risk of developing multiple sclerosis.52,53 It has been suggested that periphlebitis is a marker for disease activity,50,54 but this has not been confirmed.55 Vascular sheathing can occur in other diseases such as sarcoidosis, systemic lupus erythematosus, Wegener’s granulomatosis, and Behçet’s disease.56 Multiple sclerosis can be differentiated from sarcoidosis inasmuch as it is not associated with choroidal lesions.
Uveitis
Studies of records of patients at multiple sclerosis clinics have revealed that 1% to 2% have uveitis, which is a higher frequency than in the general population.57,58 Asymptomatic uveitis has been found in a range from none59 to 18%60 of patients with multiple sclerosis. This range probably reflects the difference in disease activity of the patients in the studies. All types of uveitis are observed; one form of intermediate uveitis, pars planitis, is probably the most common,57,61,62 but this has not been found in all studies.59
There does not seem to be any correlation between the type of multiple sclerosis and uveitis or between the degree of neurological disability and the type of uveitis,57,59 although the presence of optic atrophy may be protective against uveitis.60
Visual Loss
In patients with multiple sclerosis and uveitis, visual loss cannot be assumed to result from optic neuritis, because the complications of uveitis such as macular edema63,64 and retinal vasculitis with associated neovascularization63,65 can also result in visual loss. The features that suggest a diagnosis of uveitis rather than optic neuritis include photophobia, floaters, redness of the eye, and the absence of a relative afferent pupillary deficit. Nevertheless, many patients with multiple sclerosis–associated uveitis retain useful vision if it is treated appropriately.62
Neuroretinitis
Neuroretinitis is a descriptive term for optic disc swelling and macular exudates (Fig. 24-9) that classically results from infective causes (discussed in the next section). One study has suggested that the presence of neuroretinitis indicated that the patient did not have demyelinating disease.66 Thus, neuroretinitis should be differentiated from cases of optic neuritis that manifest with papillitis. However, in a retrospective review of 35 patients with neuroretinitis, 3 patients were found to have multiple sclerosis, although they also had received interferon β therapy.67
INFECTIONS
Infectious Causes of Neuroretinitis
As discussed, the presence of a swollen disc and macular exudates in a starlike pattern is known as neuroretinitis and is classically caused by infection. However, other causes such as hypertension can result in the same clinical picture. A number of infections are classically associated with neuroretinitis and can have neurological associations. These include cat-scratch disease, Lyme disease, and syphilis. In very rare cases, toxoplasmosis manifests this appearance.68 The ocular features of these other conditions are discussed as follows, except for toxoplasmosis, which is discussed elsewhere.
Cat scratch fever is caused by Bartonella henselae. There may be an associated uveitis, and the other fundal appearances include discrete retinal or choroidal lesions, which are more common than in classic neuroretinitis.69
Lyme disease is a spirochetal infection that results from tickborne transmission of Borrelia burgdorferi. Conjunctivitis is the most common ocular manifestation of early Lyme disease and occurs in approximately 10% of cases. Uveitis is a relatively rare manifestation of Lyme disease, but it may occur in the later stages. Vitritis, choroiditis optic neuritis, and motility problems have also been described.70–74 Exudative detachment associated with the choroiditis74 has been reported, as has a case of retinal vasculitis.75
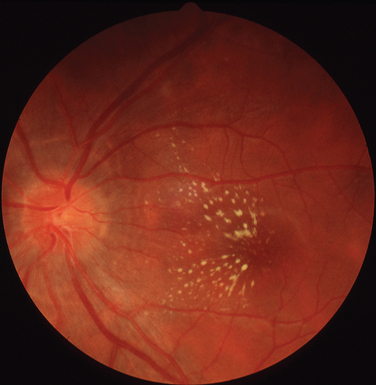
Syphilis is caused by Treponema pallidum. Between 1995 and 2003, the increase in the incidence of syphilis in the United Kingdom has been more than 10-fold.76 Ocular disease typically occurs in secondary syphilis but is also observed in tertiary syphilis. Uveitis is the most common ocular manifestation in both these stages of the disease.77,78 The retinal changes are essentially similar in both secondary and tertiary syphilis. In keeping with its moniker, the “great imitator,” the disease can affect all structures of the eye. The combination of good visual acuity, reasonable visual fields, mild panuveitis, swollen disc, and afferent papillary defect is highly suggestive of syphilitic infection.
In addition to neuroretinitis, the retinal findings include chorioretinitis, retinal vasculitis, serous retinal detachment, and necrotizing retinitis (Fig. 24-10). The necrotizing retinitis may be such that it is difficult to clinically distinguish from acute retinal necrosis (ARN) (discussed later in this chapter). The diagnosis is established by the presence of other systemic features.79 The association with HIV is discussed elsewhere in this chapter.
Tuberculosis
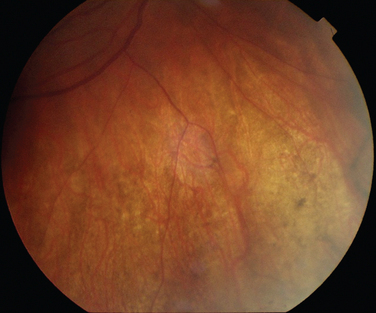
Choroidal involvement is probably the most common form of ocular involvement in tuberculosis. Usually it is in the form of choroidal tubercles that are white, gray, or yellow lesions (Fig. 24-11). The patient neither has to be seriously unwell nor has to have miliary tuberculosis for ocular disease to be present.80,81 The ocular disease may manifest many years after the tuberculosis has been found and treated elsewhere in the body.82 Other manifestations include multifocal choroiditis or serpiginous-like choroiditis.
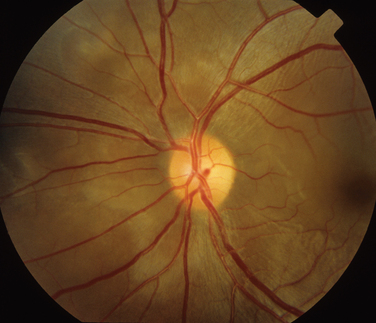
Retinal involvement can result from either choroidal extension or bloodborne spread. Retinal involvement can be either as tubercles or as a diffuse retinitis. Neovascularization and retinal vasculitis can also occur.83,84
Whipple’s Disease
Whipple’s disease is characterized by arthralgia, abdominal pain, and weight loss. The causative organism is Tropheryma whippelii. Oculomasticatory myorrhythmia is believed to be pathognomonic, but fundal changes also occur. These include vitreous opacities, diffuse chorioretinal inflammation with capillary involvement, and retinitis.85–89
Herpetic Infections
Herpesviruses cause ARN. The clinical characteristics for the diagnosis are focal, well-demarcated areas of retinal necrosis, which progresses in a rapid and circumferential manner; occlusive vasculopathy; and a prominent inflammatory reaction (Fig. 24-12).90 ARN may occur in isolation or in association with other forms of herpes infection, such as herpes zoster ophthalmicus. ARN may also be present in association with either meningitis or encephalitis. It appears that encephalitis, either concurrent or past, is more likely to be associated with herpes simplex virus type 1 infection, whereas herpes simplex virus type 2 is more likely to be involved in the presence of meningitis, whether past or concurrent.91 Genotypical analysis in two patients who had ARN after encephalitis revealed identical strains of herpes simplex virus type 1 in the brain and eye, suggestive of brain-to-eye transmission of the infection in these patients.92 ARN may occur concurrently with encephalitis or up to 20 years later.93 A case of cerebral vasculitis in which ARN was the only manifestation of any herpetic infection has been reported.94
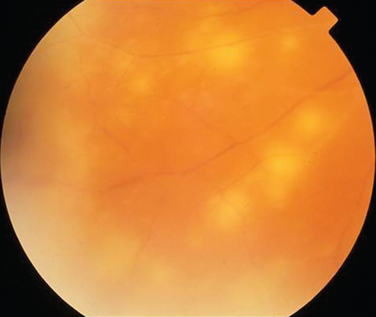
Subacute Sclerosing Panencephalitis
Subacute sclerosing panencephalitis can occur months to years after measles infection. It usually affects children or young adults, but a case in a 49-year-old man has been described.95 Ocular involvement occurs in about 50% of cases and most often develops at the same time as neurological symptoms. Chorioretinitis with subsequent scarring is the most common retinal appearance. In rare cases, the ocular signs precede the neurological ones.96
Human Immunodeficiency Virus
The retinal manifestations of HIV can be subdivided into those resulting primarily from the HIV infection, from associated infections, and from malignancies. The incidence of many of these conditions appears to have decreased since the introduction of highly active antiretroviral therapy.97 Diagnosis can be difficult because the ocular signs may differ from those in an immunocompetent individual, and there may be two concurrent infections. Also interesting is that although HIV has contributed to the increase in incidence of tuberculosis, this may not have been mirrored by an increase of ocular tuberculosis. An autopsy study of eyes from 235 patients with HIV infection revealed intraocular tuberculosis in only two eyes.98
Retinal Manifestations of HIV Infection
Microvasculopathy is the most common ocular manifestation of HIV infection, being present in 40% to 60% of patients,99 and appears to be inversely related to the CD4 count.100 In the retina, it is most frequently represented by cotton-wool spots, which are usually asymptomatic and resolve after about 6 weeks. Hemorrhages may occur, particularly in patients with low platelet counts or diabetes. They are a response to the HIV infection. Less commonly, patients may develop macular ischemia,101 which is also probably caused by the primary infection. Large retinal vessel disease has been described,102 but this is rare. Progressive visual loss has been described in a patient with HIV infection who had a clinically normal retina but abnormal photoreceptor function on electrophysiological testing.103 This process continued even when the viral load was undetectable.
Cytomegalovirus-Associated Retinitis
Cytomegalovirus-associated retinitis is the commonest ocular infection in patients with acquired immunodeficiency syndrome (Fig. 24-7). However, its incidence has dropped since the introduction of highly active antiretroviral therapy.104 It rarely occurs in patients with a CD4 count lower than 50.100 Cytomegalovirus-associated retinitis occurs mainly in patients with HIV infection but can occur in patients who are immunocompromised for other reasons.41,105 There are two main forms of clinical presentation: a fulminant form and an indolent form. The fulminant form is characterized by confluent retinal necrosis with hemorrhage that in most cases develops in the posterior retina, whereas the indolent form has a more granular form. Up to 15% of patients with cytomegalovirus-associated retinitis are asymptomatic; therefore, screening is required for patients with HIV infection who have low CD4 counts and positive cytomegalovirus serological profiles.
Herpetic Infections Associated with HIV Infection
Necrotizing herpetic retinopathy represents a continuum of posterior segment inflammation caused by herpesviruses. The best recognized are ARN and progressive outer retinal necrosis. Usually ARN occurs in immunocompetent patients or in patients with HIV infection but minimal immune dysfunction; progressive outer retinal necrosis occurs in patients with HIV and significant immune compromise. Progressive outer retinal necrosis consists of a retinitis, often in the posterior pole, that is not usually associated with an inflammatory reaction. However, the two diseases can occur simultaneously, one eye having ARN and the other progressive outer retinal necrosis.106 Both necessitate intraocular and systemic antiviral treatment.
Syphilis in HIV Infection
Ocular syphilis has been discussed elsewhere, but it has a number of significant features in relation to HIV infection. It can develop when CD4 counts are higher than 200 and can be the manifesting sign of HIV infection. The ocular findings are similar to those in patients without HIV infection, but a rare appearance, believed to be more common in patients infected with both HIV and T. pallidum, has been described. This is a placoid chorioretinopathy, consisting of large, yellowish placoid lesions with faded centers in the region of the macula and optic disc.107 There are two important features about syphilis infections in HIV-seropositive individuals. First, it can mimic the appearance of other ocular conditions such as cytomegalovirus-associated retinitis, making diagnosis and treatment difficult. Second, ocular syphilis is associated with CNS involvement in 85% to 100% of patients with HIV infection, as opposed to 35% to 40% of HIV-seronegative patients.108,109
Toxoplasmosis in HIV Infection
Toxoplasmosis retinitis can be similar to cytomegalovirus-associated retinitis, but there is usually more inflammation, and it is less likely to be associated with hemorrhage. The classic appearance in an immunocompetent patient is that of a fluffy white chorioretinal lesion in association with an area of scarring in one eye. However, in immunosuppressed patients, it can be bilateral, multifocal, and not associated with an old scar (Fig. 24-13). All of these features are suggestive of a primary infection rather than a reactivation of the condition. More than 50% of patients with ocular toxoplasmosis may have simultaneous toxoplasmosis cerebritis.99 Other fundal appearances include choroiditis, numerous scattered white lesions (a “miliary” pattern),110 a diffuse necrotizing retinitis,111,112 and punctate lesions in the outer retina.113
Malignancies Associated with HIV Infection
In patients with HIV infection, ocular non-Hodgkin lymphoma is usually associated with CNS and systemic involvement. The fundal features include necrotizing retinitis, choroidal infiltrates, and vitritis, but these appearances can also be as a result of infection by syphilis, toxoplasmosis, or viruses.114–118 Therefore, the diagnosis must be considered if the retinitis is unresponsive to antiviral, antisyphylis, and antitoxoplasmosis treatment.
Fungal Infections
A number of fungi can affect the brain and the retina. These are most often present in patients who are immunocompromised. The incidence of ocular fungal infections, such as Pneumocystis and Cryptococcus organisms, in HIV-seropositive patients appears to have decreased since the introduction of highly active antiretroviral therapy.97
Mucormycosis
Mucormycosis is an acute fungal infection that is rapidly invasive and carries a high mortality rate. Diabetes is a common predisposing factor. There are a number of clinical forms of mucormycosis, the most common being rhino-orbito-cerebral. This form affects the eye, and a necrotic eschar of the nose or hard palate is a characteristic sign. Although orbital cellulitis is the most common form of ocular involvement, serous retinal detachment and retinitis119 and choroidal ischemia have been described.120,121
Cryptococcus
Cryptococcal infection occurs mainly in patients who are HIV-seropositive. Up to 25% of patients with cryptococcal meningitis have neuro-ophthalmological lesions, which makes it the most common cause of acquired immunodeficiency syndrome–related neuro-ophthalmological lesions.99 Cryptococcal choroiditis is, however, rare. It may be may be multifocal, solitary, or confluent (Fig. 24-14).122 The lesions may initially be asymptomatic. Progressive visual loss may occur as a result of papilledema and also fungal optic nerve sheath invasion.123
Pneumocystis Carinii
One of the ocular features of P. carinii is choroiditis.124 It is classically bilateral and multifocal. The lesions are distinctive, being yellowish and well demarcated. They are slowly progressive, and usually vision is unaffected. There is not usually any associated ocular inflammation.124,125 In HIV-seropositive patients with presumed P. carinii choroidopathy, the majority had used inhaled pentamidine as prophylaxis against recurrent Pneumocystis-related pneumonia.125
MITOCHONDRIAL DISEASES
Pigmentary changes in the retina may occur in patients with mitochondrial disease. These can take a variety of forms and include appearances that have been described as pigment clumping, atrophy, salt-and-pepper retinopathy, and appearances of classic retinitis pigmentosa (Fig. 24-15). Involvement of the macula can occur, as can vascular attenuation. The most common appearance is that of a salt-and-pepper retinopathy. There does not, however, appear to be any link between the presence or type of retinal pigmentary change, the type of genetic defect, biochemical abnormality, and any clinical features.
A pigmentary retinopathy is a major diagnostic criterion for the diagnosis of Kearns-Sayre syndrome (the other features being a chronic progressive external ophthalmoplegia; onset before the age of 20; and cardiac conduction abnormalities, elevated cerebrospinal fluid protein levels, or cerebellar dysfunction). Retinal pigmentary degeneration can also be present in patients with chronic progressive external ophthalmoplegia and no other neurological or systemic abnormalities, as well as in otherwise unaffected relatives.126
Pigmentary retinopathy is also a major diagnostic feature of the syndrome of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP). However, patients who have no retinopathy, no subtle pigmentary retinopathy, and no severe bone spicule pattern have been identified with the same mutation.126 Furthermore, a patient in whom the retinal changes progressed from an initial salt-and-pepper retinopathy to typical retinitis pigmentosa over an 8-year period has been described.127
Other mitochondrial diseases in which retinal changes have been reported include the syndrome of myoclonic epilepsy and ragged red fibers and the syndrome of mitochondrial encephalopathy, lactic acidosis, and strokelike episodes (MELAS). The retinal changes in MELAS are in the form of pigmentary changes in the macula.128,129 MELAS is associated with the 3243 mutation, which can also manifest as maternally inherited diabetes and deafness. Macular changes similar to those in MELAS have also been reported in these patients,130,131 which demonstrates the marked overlap between clinical syndromes.
Funduscopic abnormalities may be present in patients with Leber’s hereditary optic neuropathy and in their asymptomatic maternal relatives. Especially during the acute phase of visual loss, there may be hyperemia of the optic nerve head, dilation and tortuosity of vessels, hemorrhages, circumpapillary telangiectatic microangiopathy, or circumpapillary nerve fiber layer swelling (pseudoedema). There may be cupping of the optic disc and arterial attenuation.126 In 2 of 20 patients with Leber’s hereditary optic neuropathy, pigmentary changes were noted at the retina.128
NEOPLASIA
Metastatic Disease
Metastases are probably the most common form of intraocular malignancy and can be associated with concurrent neurological involvement. They tend to involve the choroid but can affect the retina or vitreous humor (Fig. 24-16).132,133 Lung and breast carcinoma are the most common sources of ocular metastases and may be the manifesting sign of the disease.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


