Fig. 20.1
Satellite glial cells (SGCs), shown by confocal microscopy. A single SGC (*) was injected with the fluorescent dye Lucifer yellow, and the dye passed via gap junctions to neighboring SGCs that make an envelope around an unlabeled sensory neuron (center). Some of the SGCs are indicated with arrows. Guinea-pig DRG. Calibration bar, 50 µm
20.2 Satellite Glial Cells in Sensory Ganglia
Traditionally, pain therapy focuses on neurons (in particular in the spinal cord), but has been argued that the frequent failure in the treatment of chronic pain is due to targeting the neurons, whereas a more suitable target might be glial cells (Milligan and Watkins 2009) . Indeed, there is strong evidence that microglia and astrocytes in the spinal cord are major contributors to chronic pain (Inoue and Tsuda 2012; Ji et al. 2013) . Less is known regarding glial cells in sensory ganglia , but in light of the importance of sensory ganglia in pain, it is reasonable to assume that these cells, like their spinal cord counterparts, i.e. astrocytes, play a role in pain. The main type of glial cells in sensory ganglia are SGCs, which share many properties with astrocytes, including the expression of glutamine synthetase and various neurotransmitter transporters (for reviews see Hanani 2005; Huang et al. 2013; Jasmin et al. 2010; Takeda et al. 2009) . A unique feature of SGCs, which distinguishes them from astrocytes, is that several of them usually form a sheath that surrounds individual sensory neurons completely, thus forming a neuron-SGC functional unit (Hanani 2005; Hanani and Spray 2013; Pannese 2010) (Fig. 20.1). SGCs within this unit are coupled by gap junctions (Cherkas et al. 2004; Hanani et al. 2002) . The pharmacology of SGCs is largely unexplored, but it is known that they are endowed with functional receptors for ATP (Ceruti et al. 2008; Huang et al. 2013; Magni and Ceruti 2013; Weick et al. 2003) , cytokines (Huang et al. 2013; Takeda et al. 2009) , and endothelin (Feldman-Gorianchik and Hanani 2011) . The presence of gap junctions and purinergic P2 receptors that respond to ATP is noteworthy as both have been shown to mediate the spread of intercellular calcium waves between astrocytes (Scemes and Giaume 2006) and also in SGCs (Suadicani et al. 2010). SGCs release ATP (Huang et al. 2010; Suadicani et al. 2010) and cytokines (Takeda et al. 2009; Zhang et al. 2007) and possibly other chemical messengers, which enable bidirectional communication between these cells and neurons. SGCs undergo major changes as a result of injury to peripheral nerves, and appear to contribute to chronic pain in a number of animal pain models (Dublin and Hanani 2007; Huang et al. 2010; Ohara et al. 2009; Thalakoti et al. 2007; Takeda et al. 2009) .
20.3 Survey on Changes in SGCs After Injury
Tables 20.1, 20.2, 20.3 list changes that various injuries induce in SGCs in sensory ganglia . Although comprehensive, the tables are not complete, and for older literature the reader is referred to previous reviews (Hanani 2005; Pannese 1981) . The main message of these tables is that a wide range of injury types leads to consistent effects in these cells. It should be emphasized that following injury, sensory neurons undergo major changes as well, but these are not included in the tables (for reviews see Devor 2006; Dib-Hajj et al. 2010) . It is evident from the tables that: (1) Most of our knowledge on SGCs derives from work on rats and mice. These animals are indispensable for basic research, but information on higher species, and especially humans, is missing. As rodent pain models may not exactly reflect human disease, the conclusions from rodent work should be regarded with caution when attempting to extrapolate the conclusions to humans. (2) There is a clear pattern of changes in SGCs across the various models, which include somatic, orofacial, visceral pain, and pain in systemic disease. The most prominent change is the augmented expression of GFAP, which is a well-established marker for astrocyte activation (Pekny and Nilsson 2005) . GFAP immunostaining is particularly useful in the case of SGCs, because under resting conditions, GFAP level is very low, whereas it is greatly augmented after injury. GFAP belongs to the family of intermediate filament proteins, and has structural roles in astrocytes; however, the functional significance of its upregulation its obscure (for reviews see Gao and Ji 2010; Middeldorp and Hol 2011) . It has been proposed that GFAP upregulation contributes to pain, but firm information is lacking. GFAP-null mice displayed higher degree of neuronal damage than normal mice, suggesting that GFAP has a protective role. A central question is whether GFAP is essential for glial activation, or whether it just accompanies this process. Chudler et al. (1997) commented that although changes in GFAP immunoreactivity in the TG and alterations in neuronal response properties were correlated, the causal relation between these phenomena is still unknown.
Table 20.1
Changes in satellite glial cells in DRG following somatic injury
Type of injury | Changes in SGCs | Comment | References |
|---|---|---|---|
Sciatic N axotomy | GFAP, gap junction upregulation | Rats, mice | |
Sciatic N neuritis | Dye coupling increase, gap junctions increase | Mice | Ledda et al. (2009) |
Skin scarification | Proliferation | Mice | Elson et al. (2004) |
Sciatic N axotomy | Hypertrophy (requires months to develop), proliferation | Rats | Shinder et al. (1999) |
Chronic DRG compression | Increased coupling, GFAP upregulation, reduced Kir currents | Rats | Zhang et al. (2009) |
DRG inflammation | GFAP and cytokine upregulation | Rats | Siemionow et al. (2009) |
Tetanic sciatic N stimulation | GFAP, P2 × 7 receptor upregulation | Rats | Liang et al. (2010) |
Spine injury | GFAP and TNF-alpha upregulation | Rats | Miyagi et al. (2006); |
Spinal N crush | GFAP and TNF-alpha upregulation | Rats; ipsi- and contralateral DRG affected | Hatashita et al. (2008) |
Non-compressive disc herniation | GFAP upregulation | Rats; ipsi- and contralateral DRG affected | Li et al. (2011a) |
Intervertebral disc injury | GFAP upregulation | Rats | Li et al. (2011b) |
Plantar skin incision | GFAP upregulation | Mice | Romero et al. (2013) |
Paw inflammation | P2 × 7 receptor upregulation | Rats | Chen et al. (2008) |
Paw inflammation | Dye coupling increase | Mice | Dublin and Hanani (2007) |
Sciatic N CCI | IL-6 upregulation | Rats, ipsi- and contralateral DRG affected | Dubový et al. (2010) |
Periganglionic inflammation | COX-2 upregulation | Rats; | Amaya et al. (2009) |
Sciatic N ligation, root sections | p75 upregulation | Rats | Zhou et al. (2001) |
Sciatic N axotomy | FGF-2 and S100β upregulation | Rats | De-Freitas Azvedo Levy et al. (2007) |
Sciatic N crush | FGF-2 upregulation | Rats | Grothe et al. (1997) |
Mild sciatic N compression | GFAP upregulation, macrophage infiltration | Rats; carpal tunnel syndrome model | Schmid et al. (2013) |
Sciatic N crush | TNF receptor, TNF upregulation | Rats | Ohtori et al. (2004) |
Trauma | P2 × 7 receptors upregulation | Human | Chessell et al. (2005) |
Sciatic N. CCI | Cannabinoid type 2 receptor upregulation | Rats; ipsi- and contralateral DRG affected | Svízenská et al. (2013) |
Spinal N ligation | GFAP upregulation | Rats | |
Spinal N ligation | MMP-2 upregulation | Rats | Kawasaki et al. (2008) |
Spinal N ligation | HMGB-1 upregulation | Rats | Shibasaki et al. (2010) |
Spared nerve injury | GFAP upregulation | Rats | Xie et al. (2009) |
Bone cancer | GFAP upregulation | Mice | Peters et al. (2005) |
Table 20.2
Changes in satellite glial cells in TG following orofacial injury
Type of injury | Changes in SGCs | Comment | References |
|---|---|---|---|
Temporo-mandibular joint inflammation | Connexins 26,36,40 upregulation | Rats | Garrett and Durham (2009) |
Capsaicin in temporo-mandibular joint | S100B and p38 expression increased | Rats; all divisions of TG affected | Thalakoti et al. (2007) |
Submanibular inflammation | P2 receptors upregulation | Mice; calcium imaging study | Kushnir et al. (2011) |
Whisker pad inflammation | Kir1.4 downregulation | Rats | Takeda et al. (2011) |
Whisker pad inflammation | GFAP, IL-1β upregulation | Rats | Takeda et al. (2007) |
Infraorbital N CCI | Connexin 43 upregulation | Rats | Ohara et al. (2008) |
Infraorbital N CCI | Kir1.4 downregulation | Rats | Vit et al. (2008) |
Infraorbital N CCI | Proliferation | Rats | Donegan et al. (2013) |
Single tooth injury | GFAP upregulation in all parts of TG | Rats | Stephenson and Byers (1995) |
Inferior alveloar N crush | GFAP upregulation | Rat | Chudler et al. (1997) |
Partial infraorbital N ligation | Proliferation, GFAP upregulation | Mice | Xu et al. (2008) |
Infraorbital N axotomy | Dye coupling increase, sensitivity to ATP increased | Mice | Cherkas et al. (2004) |
Temporo-mandibular joint inflammation | ERK phosphorylation, | Rats | Bi et al. (2013) |
Temporo-mandibular joint inflammation | GFAP upregulation | Rats; all divisions of TG affected | Villa et al. (2010) |
Infraorbital N axotomy | P2 receptor upregulation | Mice; calcium imaging study | Kushnir et al. (2011) |
Tooth extraction | GFAP upregulation | Rats | Gunjigake et al. (2009) |
Tooth pulp inflammation | GFAP upregulation | Rats | Matsuura et al. (2013) |
Tooth movement | NGF upregulation | Rats | Kurata et al. (2013) |
Lingual N crush | GFAP, P2Y12R upregulation | Rats; all divisions of TG affected | Katagiri et al. (2012) |
Table 20.3
Changes in satellite glial cells in DRG or TG following miscellaneous types of injury
Type of injury | Changes SGCs | Comment | References |
|---|---|---|---|
Chemotherapeutic drugs (systemic) | Dye coupling increase, GFAP upregulation | Mice DRG | |
Systemic LPS | Dye coupling increase, GFAP upregulation | Mice DRG | Blum and Hanani unpublished |
Water avoidance stress | GFAP upregulation | Rat DRG | Golovatscka et al. (2012) |
Aging | Dye coupling increased | Mouse DRG | Huang et al. (2006) |
Intestinal inflammation or obstruction | Dye coupling increased | Mouse DRG | |
In vitro bradykinin | P2Y receptor upregulation | Cultured mouse TG | Ceruti et al. (2008) |
Diabetes model | Aldose reductase upregulation | Rats | Jiang et al. (2006) |
Acute morphine | GFAP, IL-1β upregulation | Mice; acute increases | Berta et al. (2012) |
Simian immunodeficiency virus | Virus infection | Monkeys; a model for HIV | Burdo et al. (2012) |
Antiretroviral drug | Chemokine upregulation | Rats | Bhangoo et al. (2007) |
Another change in SGCs after a variety of injuries in mice is the increase in gap junctions among SGCs (Figs. 20.2 and 20.3). Much less work has been done on rats, and the only dye coupling study in rats was done on rat DRG (Zhang et al. 2009) . There is also immunohistochemical evidence for an increase in connexins expression in rat TG after damage (Garrett and Durham 2009; Ohara et al. 2008) . Increased cytokine synthesis is a common feature in SGCs following damage, which may have functional significance, as these mediators can contribute to neuronal excitation.
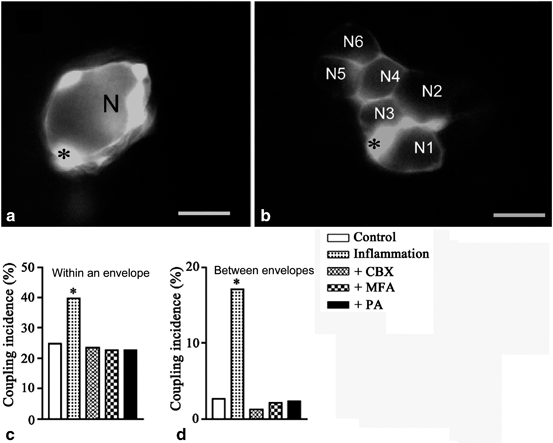
Fig. 20.2
Dye coupling in sensory ganglia is augmented by peripheral inflammation. The fluorescent dye Lucifer yellow (LY) was injected into SGCs of S1 DRG ganglia of mice to determine the degree of gap junction mediated coupling between the cells. Dinitrobenzoate sulfate (DNBS) was instilled into the large intestine to induce inflammation, and the ganglia were examined in vitro 10–12 days later. a LY-injected SGC is coupled to other SGCs only around the same neuron. b Dye coupling between SGCs around different neurons (N1-N5), observed in DNBS-treated mouse. The asterisks indicate the LY-injected SGCs. Scale bars, 20 µm. The histograms (c, d) show the effect of gap junction blockers: carbenoxolone (CBX, 50 µM), meclofenamic acid (MFA, 100 µM) and palmitoleic acid (PA, 30 µM), on the augmented coupling among SGCs after inflammation. c Incidence of coupling between SGCs around the same neuron. d Incidence of coupling between SGCs around different neurons; N = 81 ± 129 for each of the experimental conditions, p < 0.001 compared with results obtained after DNBS in the absence of blockers. *p < 0.01, N = 17 ± 29. Fisher’s exact test. (Modified from Huang et al. 2010)
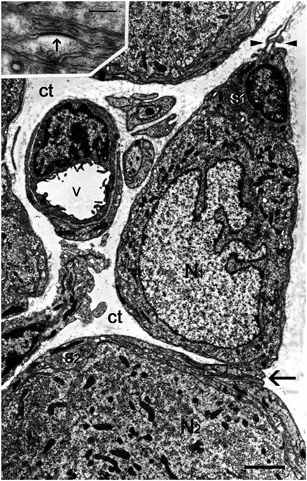
Fig. 20.3
Electron micrograph showing the effect of peripheral inflammation on SGCs. Neuritis of the sciatic nerve caused the formation of a bridge (arrow) connecting two SGCs (s1, s2), each part of the sheath around a different neuron (N1, N2). The inset is a high magnification of the boxed area, which is within the bridge formed by the two SGCs. The dark band (indicated by an arrow) is a gap junction. On the basis this micrograph alone it cannot be determined that the gap junction is between two different SGCs, because the gap junction can be between different processes of the same SGC. However, the dye coupling results strongly support the conclusion that injury increases SGC-SGC coupling by gap junctions. Minute protrusions (arrowheads) emerge from the outer contour of the SGC sheath (s1) of the neuron N1 and project into the connective tissue space (ct). v, blood vessel. Scale bar, 2 µm; inset: scale bar, 0.2 µm
Our knowledge on the pharmacology of SGCs is limited, particularly in pathological states. SGCs display P2X and P2Y purinergic receptors . In two injury models in mice, it was found that the sensitivity of SGCs to ATP is increased 100-fold (Kushnir et al. 2011) . In one of these models (submandibular inflammation) the P2 receptor population appears to switch from P2Y to P2X. Following in vitro inflammation, P2Y receptors are upregulated in SGCs (Ceruti et al. 2008) . Katagiri et al. (2012) showed an increase in P2Y12 receptors in rat following lingual inflammation. All these studies were done in TG, and there is only a preliminary study showing an increase in responses to ATP in mouse DRG due to systemic inflammation (Hanani 2013). As SGCs are likely to communicate among themselves and also with neurons by chemical messengers, much more work needs to be done on pharmacological changes in SGCs. Although the understanding of SGCs under normal and pathological conditions is rather limited, the available information provides several promising leads to possible pain therapies that could be based on SGC-neuron interactions. Several recent review articles on pain therapy present a rather deplorable situation: there is an extremely wide range of pain types, and there are dozens of mechanisms that seem to be involved (Gold and Gebhart 2010; Ren and Dubner 2010) . For example, several types of voltage sensitive Na+ channel are found in sensory neurons, and different human pain disorders are associated with an increase in different Na+ channels (e.g., Nav1.3), but paradoxically in some cases Na+ channels are downregulated after injury (e.g., Nav1.7 and 1.8) (Berta et al. 2008; Dib-Hajj et al. 2010) . For these types of Na+ channels, evolution has favored mutations in certain rodents rendering Nav1.7 in the mole rat insensitive to acid (Smith et al. 2011) and Nav1.8 in the grasshopper mouse being inhibited rather than excited by scorpion toxin (Rowe et al. 2013) . Numerous biochemical pathways are altered in pain states, and various studies emphasize a given pathway as being central to the pain syndromes, with the exclusion of others. The conclusion of the reviews is that there is little hope of making substantial progress in this field. These conclusions might be indeed justified, but perhaps we should look at this problem from a fresh angle. For example, there is growing evidence that glial cells in the spinal cord can serve as a therapeutic target (Ji et al. 2013) . However, targeting the CNS has several limitations, the main one being the presence of the blood brain barrier, which limits the types of drugs that can be used. Secondly, the great structural and pharmacological complexity of the CNS is a serious obstacle. In contrast to the CNS, sensory ganglia are rather simple structurally, and display a very limited neuronal and glial diversity. These ganglia offer additional advantages: there is no blood-ganglion barrier, and as they are an early station in the pain pathways, targeting them is more efficient, as it can prevent the development of central changes.
20.4 Selecting Therapeutic Targets in SGCs
Astrocytes usually share the neurotransmitters and receptors of their neighboring neurons (Verkhrarsky 2009) . This seems to hold also for SGCs. Thus, it is not easy to target SGCs chemically without also influencing the neurons. Still, some pharmacological differences between SGC and sensory neurons have been found and may be utilized to target SGCs selectively. Another strategy is to aim at abnormal SGC-neuron interactions. Below we list several such possibilities.
20.4.1 Potassium Channels
The main K+ channel in astrocytes is the inward rectifying (Kir)4.1 channel, which plays a key role in generating the membrane potential and in regulating the extracellular K+ concentration, i.e. “K+ spatial siphoning/buffering”. K+ that accumulates outside neurons during neuronal activity enters the astrocytes via Kir4.1 channels and is distributed through the cell (siphoning) or glial syncytium (buffering) to regions where K+ concentration is low, thus reducing the danger of excessive neuronal depolarization. These channels are ideally suited for this function because their conductance is larger in the inward than the outward direction and increases when the external K+ concentration increases. We first identified Kir channels in SGCs from mouse TG (Cherkas et al. 2004) , which were later identified as predominantly Kir4.1 (Takeda et al. 2011; Tang et al. 2010; Vit et al. 2008) , and were found to be the most abundant channels in SGCs. Although direct evidence for K+ buffering in SGC is still lacking, it is highly likely that SGCs, like astrocytes, can regulate the external K+ concentration. Several studies have shown that Kir4.1 are downregulated in sensory ganglia following damage (Takeda et al. 2011; Vit et al. 2008), which is expected to lead to K+ accumulation in the extracellular space and to neuronal depolarization, causing increased neuronal firing and possibly neuronal damage. It is noteworthy that Kir4.1 is found only in SGCs and not in sensory neurons. Moreover, it was found that these channels are downregulated in TG in two different pain models. In support of this, silencing of Kir4.1 using RNA interference led to spontaneous and evoked facial pain-like behavior in rats (Vit et al. 2008) . Therefore manipulations that open these channels have a clear therapeutic value. To our best knowledge currently there is no compound that can open Kir4.1 channel, and a search for such a drug holds considerable promise.
Related posts:
 Experimental Treatment of Acquired and Inherited Neuropathies
Possible Therapeutic Targets in Microglia
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
Amyotrophic Lateral Sclerosis: A Glial Perspective
Enteric Glial Cells: Implications in Gut Pathology
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Experimental Treatment of Acquired and Inherited Neuropathies
Possible Therapeutic Targets in Microglia
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
Amyotrophic Lateral Sclerosis: A Glial Perspective
Enteric Glial Cells: Implications in Gut Pathology
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


