Figure 36.1. The cerebral blood flow (CBF)-pressure curve.
This phenomenon is generally seen under normal physiologic conditions across a blood pressure range of 50-150 mmHg; however, longstanding hypertension causes a “right shift” of the autoregulatory curve. Failure of pressure autoregulation typically occurs outside this blood pressure range, and there is strong evidence that it may become dysfunctional after TBI [23,24].
Metabolic autoregulation is represented by the Fick equation:
CMRO2= CBF x AVO2D
Because the cerebral metabolic rate, cerebral blood flow, and arteriovenous oxygen difference are all interconnected, the brain is able to precisely match local cerebral blood flow to local metabolic needs. The exact cellular mechanisms behind pressure and metabolic autoregulation are not completely understood, but there is evidence that the vascular endothelium has an integral role. Studies have shown that an intact vascular endothelium is essential for maintaining cerebrovascular hemostasis, largely through local metabolites and vasoactive substances such as H+, CO2, adenosine, K+, calcium, nitric oxide, endothelin and thromboxane [25]. All these substances have all been proposed as possible mediators of cerebrovascular reactivity.
Cerebral blood flow may be accurately measured in the intensive care unit (ICU) using xenon gas, the 133Xe method, in xenon-enhanced computed tomography (CT). However, radiation exposure and costs limit the frequency of its use to 2-3 scans per patient. With the recent introduction of the Hemedex continuous CBF monitor based on a thermal diffusion method, both blood supply and metabolism can be measured in the ICU.
36.5 Secondary Brain Injury
Focal and global alterations in cerebral metabolism, cerebral blood flow, autoregulation, and intracranial pressure contribute to the development of secondary brain injury. The cerebral metabolic rate in head-injured patients is typically reduced, ATP generation is lower, and anaerobic glycolysis is critical under circumstances of severely reduced CBF or mitochondrial damage [26]. This results in increased lactate concentration and acidosis. Disturbances in autoregulation, or “dysautoregulation,” a mismatch of cerebral blood flow and metabolism, and impaired CO2 reactivity may occur together and have a harmful effect on the injured brain. These events, coupled with systemic perturbations, such as hypoxia and hypotension due to lung injury, airway obstruction or shock, can combine to wreak havoc on potentially salvageable neurons. Ischemic brain damage is very common after severe head injury, being demonstrated in up to 80% of autopsy subjects [27].
The brain’s initial response to ischemia or declining blood flow is to extract escalating amounts of oxygen from the circulation. But when extraction reaches a maximum, further reductions in blood flow result in energy failure, mitochondrial and cell membrane disruption, cell swelling and cell death [28,29]. The ultimate determination of cell survivability versus cell death depends on a complex relationship between actual blood flow, duration and degree of ischemia, specific cell type, glucose concentration and temperature, among other factors. There seems to be a relatively constant value of blood flow associated with salvageable ischemic neurons and irreversible cell death: 18 cc/100 g/min and 8 cc/100 g/min, respectively [29].
Measuring cerebral perfusion pressure (CPP) is an indirect way to assess CBF in real time. Cerebral blood flow is related to intracranial pressure (ICP) and mean arterial pressure (MAP) by the equation:
CPP = MAP – ICP
Increased ICP is a common event contributing to secondary brain injury in severely injured patients, and it is often the main clinical concern that determines treatment.
36.6 Intracranial Pressure
The Monro-Kellie hypothesis states that the cranium is a rigid non-expandable box and that its contents consist of mainly three volumes: the brain (80%); cerebrospinal fluid (CSF) (10%); and blood (10%). In order for the pressure inside this rigid box to remain constant, the volumes of the contents must remain fairly stable. Any increase in volume of the original contents (hyperemia/edema) or owing to a mass-occupying lesion (hematoma) must be offset by a decrease in volume of one of the other components, otherwise the ICP will rise. The compensatory mechanism begins with the diversion of CSF and intracranial blood. Over 80% of severely head-injured patients have persistently elevated ICP [30]. In the healthy adult the ICP is 5-15 mmHg, and it is usually 0-10 mmHg in children. Persistently elevated ICP has been associated with poor outcomes, and mortality is directly related to the degree and duration of elevated ICP [31,32].
As ICP rises and compensatory mechanisms reach their limit, CPP generally decreases, setting the stage for decreased blood flow, ischemia, and neuronal cell death. Much of the clinical care in the neuro-ICU is directed at preventing and managing increases in ICP by maintaining adequate perfusion pressures and aggressively avoiding ischemic situations brought about by hypoxia or hypotension. Other modalities include evacuation of intracranial mass lesions and limiting cerebral edema. Specifics on monitoring and treatment strategies for raised ICP will be discussed later in this chapter.
36.7 Intracranial Hypertension, Monitoring Modalities, and Treatment Strategies
36.7.1 Intracranial Hypertension
Elevated ICP is the leading cause of death in head-injury fatalities, and it is a major contributing factor to the secondary insult mechanism of brain injury. The capacity to accommodate up to 50 cc of new intracranial volume without a significant rise in ICP occurs by the displacement of venous blood into the general circulation and of CSF out of the cranial vault into the spinal subarachnoid space; however, these mechanisms are both age- and time-dependant. Since elderly people tend to have more cerebral atrophy, they may accommodate a larger amount of a slowly expanding volume. Younger individuals with an acute process, on the other hand, are much more likely to become rapidly symptomatic. All surgical lesions should be aggressively evacuated according to Brain Trauma Foundation surgical guidelines. Mass lesions will be discussed in the subsequent section.
Abnormal cerebral autoregulation, new hemorrhage, and cerebral edema are the major causes of delayed intracranial hypertension. Clinical trials have shown that head-injured patients with an ICP >20 mmHg, particularly when sustained or intractable to treatment, have a worse clinical outcome and are at high risk for the herniation syndrome [29-33]. There is also recent evidence that CPP <60-70 mmHg is associated with worsened parenchymal oxygenation, metabolism, and outcome [34]. Therefore, the goals of monitoring and treatment are to maintain adequate CPP, oxygenation, and metabolism, while limiting elevation in ICP, edema, and substrate perturbations.
Monitoring Techniques
Monitoring techniques in the head-injured patient can be loosely classified under three categories: pressure monitoring; blood-flow monitoring (Figure 36.2); and substrate monitoring (Figure 36.3). The choice between monitoring systems is becoming increasingly complex, and the capability to monitor multiple parameters such as oxygenation and substrate concentrations, as well as ICP is becoming increasingly common. As mentioned previously, brain ischemia, hypoxia, and cellular malfunction are common following TBI, and the reduction in blood flow and substrate delivery can occur within minutes, stressing the importance of early resuscitation and management of the intracranial environment.

Figure 36.2. Cerebral blood flow monitoring device.
Figure 36.3. Substrate monitoring.
Pressure Monitoring: Types and Indications
Lundberg introduced ventricular drainage as a management strategy for treating intracranial hypertension in the 1960s. However, the concept of ICP monitoring did not gain momentum until the mid-1980s with evidence-based literature describing the subarachnoid bolt. ICP monitoring technology has grown since its inception and current ICP monitoring options include subdural, epidural, parenchymal and intraventricular monitors. Today, most monitoring is limited to parenchymal and intraventricular monitoring because subdural and epidural monitors are inaccurate. Ventriculostomy is the gold standard for the evaluation of raised ICP. An intraventricular monitor allows for reliable diagnosis and the potential for therapeutic intervention through drainage of CSF for raised ICP. Intraventricular monitoring is a low-cost, highly accurate method; however, it carries a slightly higher post-insertion malfunction rate than parenchymal monitoring. Parenchymal monitors such as the fibre optic (Integra Neuroscience-Plainsboro, NJ) and transducer tipped devices, like the microstrain gauge (Codman-Raynham, MA.) are placed in the brain parenchyma but are generally more expensive and cannot be recalibrated once inserted [35-37].
The Brain Trauma Foundation Guidelines for the Management of Severe Head Injury recommend ICP monitoring in patients with a GCS score <9 and an abnormal CT scan (blood, edema, contusion) or a GCS score <9 with a normal CT scan, and two of the following three: 1) age >40; 2) systolic blood pressure (SBP) <90 mmHg; and 3) unilateral or bilateral motor posturing. With ICP and mean arterial blood pressure (MABP) measurements, cerebral perfusion pressure can then be determined to provide an indirect assessment of CBF. The CPP value necessary for preventing ischemia varies individually, depending largely on the integrity of autoregulation but generally lies between 50 and 70 mmHg [101,102].
36.7.2 Blood Flow Monitoring
Transcranial Doppler
Transcranial Doppler (TCD) is an inexpensive non-invasive tool that provides insight into cerebral hemodynamics, and it can alert the clinician to possible harmful events. TCD relies on an ultrasonic pulse signal that is transmitted through thin areas of the skull. Flow velocity through a vessel is determined, and flow volume may be calculated by multiplying the velocity by the cross-sectional area of the vessel. Since the amount of collateral flow and the actual vessel diameters are unknown, TCD cannot provide quantitative data on regional tissue perfusion. Changes in flow velocity can, however, provide relative data on changes in flow volume. Typically, flow velocities >200 cm/second indicate severe vasospasm, vessel narrowing, and impending infarction. Hyperemia, a common occurrence in post-TBI, may also lead to elevated flow velocities. The so-called Lindegaard ratio or hemispheric index (middle cerebral artery [MCA]/extracranial internal carotid artery [ICA]) may help differentiate between the two situations, with normal values being <2 and critical values being >3 [38].
Xenon CT
Xenon CT is the most accurate non-invasive method for determining both regional and global cerebral blood flow. Xenon is a radiodense, inert and rapidly diffusible substance that allows precise quantitative measurement necessary for determining accurate blood flow values. Baseline plain CT scans are performed, followed by serial scans while the patient inhales 28-33% xenon. Complex mathematical models produce precise quantitative values, relying on Fick’s principle which states the amount of a substance taken up by an organ equals the amount delivered minus the amount carried away by venous blood. Xenon CT is not devoid of technical pitfalls: patients must be free of cardiovascular disease, both functionally and structurally, in order to produce reliable results; and if portable CT is unavailable, moving the patient to the CT scanner multiple times may also become an issue. Two invasive methods for measuring continuous local CBF are now also available and have correlated well with xenon CT techniques: the thermal diffusion method (Hemedex) and the laser Doppler method. Both methods require open craniotomy or a twist-drill hole for placement, and both measure only small focal areas of brain tissue, which may or may not be representative of the global blood flow picture. Both are under evaluation in many centres.
Near-infrared Spectroscopy
Near-infrared spectroscopy (NIRS) technology has been developed as an emerging method for monitoring cerebral oxygenation, blood flow, and blood volume in both adults and children [39,43]. The technique involves the transmission of light from an emitting source through the brain to a sensor. Some studies suggest it can monitor cerebral oxygenation, blood flow, and blood volume in both adults and children [39-43]. NIRS has received technical criticism because, while transmission of the light source works well in neonates due to their semitransparent skull and scalp, monitoring in adults can be problematic due to dilution of the brain NIR signal by extracerebral tissues such as the skull bone marrow and scalp [44,45]. The emitter and detector are separated by specified distances on the scalp with the premise that a fixed amount of transmitted, reflected, and scattered light follows an elliptical path whose depth of penetration is proportional to the distance of separation between the emitter and the detector (Figure 36.2) [45].
Cerebral oximeter (Somanetics Inc, Troy, MI, Invos Cerebral Oximeter) is one such product used to estimate blood oxygen saturation in the brain. Optical transceiver pairs are placed on the scalp, and the attenuation of light signals at two wavelengths is used to estimate regional blood oxygen saturation. Its use in both operative procedures and ICU settings have been reported. Brawanski et al. demonstrated a correlation between intraparenchymal oxygen probes and NIRS probes in 9 patients following TBI, and Kirkpatrick et al. found that NIRS probes recorded changes in cerebral oxygenation that recovered once blood flow was restored during vessel cross-clamping in carotid endarterectomy [46]. The use of NIRS in conjunction with a bolus injection of indocyanine green (ICG) has been used to measure CBF. NIRS measurements may help to identify critical clinical parameters and guide treatment directions. As it is not sufficient as a stand-alone monitoring technique, these values should be considered in context with other monitoring methods.
Laser Doppler Flowmetry
Laser Doppler flowmetry (LDF) relies on an implanted subcortical fibre optic laser probe (diameter, 0.5-1 mm) that measures the shift of reflected laser light produced by the progression of red blood cells recorded in blood perfusion units (BPU.) Laser light from one fibre is scattered within the tissue and some is scattered back to the detector probe, which is another optical fibre that collects the backscattered light from the tissue and returns it to the monitor. The light returned to the monitor undergoes signal processing, wherein the emitted and returned signals are compared to extract the Doppler shift related to moving red blood cells. A BPU is a relative units scale determined using a carefully controlled motility standard comprising a suspension of latex spheres undergoing Brownian motion. The greatest benefit of LDF is that the data are presented in real time and variations in blood flow are detectable immediately. A major drawback is that the data are a qualitative and not a quantitative analysis. When used in conjunction with xenon CT, an absolute value can be determined, with xenon acting as a calibrator for the LDF [37,47].
Thermal Dilution Flow
Thermal dilution flow (TDF) methods (Hemedex, Codman, Cambridge, MA) are based upon the conductance of small amounts of heating between two small gold electrodes, one equipped with a sensitive thermocouple sensor. CBF is proportional to heat conductance, and the values are thus absolute. The probes are usually configured for placement in the deep white matter rather than the grey matter, with correspondingly much less CBF change (e.g., normal CBF in white matter is 22 ml/100 gm/min versus 80 ml/100 gm/min in the grey cortex). The sensors are usually placed via a “bolt” immobilizing system, but they can be placed at craniotomy (e.g., for aneurysm clipping). Clinical acceptance of either of these monitoring techniques has been slow thus far perhaps due to the reporting of relative values and the questionable reliability and validity of the commercial systems [27]. The development of non-invasive methods to assess both regional and global blood flow and oxygenation together are also being developed and may one day replace LDF and TDF.
36.7.3 Oxygen and Temperature
SjvO2
Jugular venous oxygen saturation (SjvO2) monitoring involves cannulation of the internal jugular vein and advancement of the catheter into the jugular bulb (about 13 cm) cephalically to the level of C1, confirmed with lateral and/or anteroposterior plain x-ray. Jugular venous oxygen saturation sampling was first utilized in the 1930s as a promising method to detect post-traumatic changes in global cerebral oxygenation, but its subsequent use has proved to be more difficult than originally envisioned [48]. The concept of SjVO2 monitoring is based upon the fact that most (about 80%) of the cerebral venous blood drains into the internal jugular veins, so that jugular venous blood indicates brain events. The normal range of SjvO2 is 50-75%, but typically lies between 50-60% in most patients. When CBF decreases, oxygen extraction increases to compensate it, and the cerebral metabolic rate does not change initially. If this relationship continues and decreases in CBF surpass this compensatory mechanism and the supply is unable to meet demand, the metabolic rate decreases and anaerobic metabolism takes over. SjVO2 decreases <50% are associated with altered brain metabolism, and values <20% are associated with irreversible ischemic injury and poor outcome [49]. In head-injured patients, however, SjvO2 measurements are widely variable, ranging from 34 to 96% in some series [50]. Measurements of SjVO2 are obtained by draining blood from the internal jugular venous catheter. Widespread use of this monitoring method has been limited however, due to catheter migration, technical difficulties of insertion, poor positioning, calibration errors (e.g., light absorbance), and the difficulty in differentiating regional versus global brain oxygenation, leading to a reported 50% error rate [48,51-53]. Questions concerning the reliability, validity and new PtiO2 technology (see below) have limited the use of SjVO2 monitoring to a few centres.
PtiO2/PbO2
The Licox monitor is a direct means to measure brain oxygen and temperature. The probe measures 0.8 mm and is placed into brain tissue to determine local or regional oxygenation and neighbouring brain temperature depending upon the insertion site. The Licox (GMS-Integra, Kiel-Mielkendorf, Germany) system [54] is a small and easily inserted miniaturized Clark electrode with a polyethylene sheath (sampling area approximately 14 mm2), and it has proved to be an accurate measurement of partial pressure of oxygen (Figure 36.4). Diffusion of oxygen from red blood cells to the extracellular space provides the substrate measurement, and the Licox system integrates both venous and arterial oxygen tension in the measured area [55]. Conformation of sensitivity can be accomplished by altering inspired FiO2 to 100% following intracerebral placement, often referred to as an “oxygen challenge” [56]. When inserted near an area of injury or penumbra, the probe can detect localized hypoxia or evaluate global brain oxygenation. Normal brain tissue oxygenation (partial pressure of brain tissue oxygen [PbtO2]) is 20-35 mmHg. Current Brain Trauma Foundation guidelines indicate a PtiO2 of 15 mmHg. Values <10-14 mmHg should be considered a sign of tissue hypoxia [35,52] and values <5-10 mmHg indicate impending neuronal death and infarction.
In early trials, Robertson and Valadka et al. demonstrated that when pO2 was maintained at normal values, it was a predictor of better outcomes in TBI patients. Bard et al. later showed some correlation with duration of low PtiO2 and poor outcome. Van den Brink et al. looked at 101 patients with non-penetrating head injury and a GCS score ≤8 and found that the magnitude and length of time of low PtiO2, as measured with the Licox system, was an independent predictor of unfavourable outcome and death. The authors noted that the device demonstrated little zero drift and that the values were reliable over the time course of monitoring [57].
Placement location remains a complicated issue to resolve. Studies have shown that direct placement of the catheter in contused brain provides inaccurate responses following increased FiO2, suggesting that CBF may be inadequate to provide oxygen delivery in these contused areas [58]. Accordingly, some groups recommend placement in non-contused white matter of the frontal lobe in diffuse injury or on the affected side in a unilateral injury [54,58]. Although these measurements may help to identify critical clinical parameters and guide treatment directions, most authors agree that PO2 monitoring is not sufficient as a stand-alone monitoring technique [55]. These values should be taken in context with other monitoring methods and should help guide rather than dictate clinical action. The risk of infection or catheter-related hemorrhage is minimal. A randomized clinical trial of PtiO2 versus non-PtiO2 monitoring-guided therapy is currently underway in the United States (the “BOOST” study). The Licox probe also has an incorporated thermocouple allowing for brain temperature monitoring.
Hyperthermia in human and animal studies has proved to be another secondary insult resulting in poor outcomes [59]. Cerebral temperature typically differs from core temperature. Schwab et al. cite 1.5°C ± 0.3. A growing body of evidence in both animals and humans suggests that induction of mild hypothermia (32-35°C) early after injury may arrest or slow secondary brain damage mechanisms.

Figure 36.4. Licox system.
Microdialysis
The cerebral microenvironment changes rapidly after TBI. Excitatory amino acid release, calcium influx, pump and membrane failure, and lactate accumulation are just a few of the derangements. Many of these changes can be detected with the use of microdialysis in humans. Correlations between adverse clinical events (e.g., elevated ICP, hypotension or hypoxia) and increased dialysate concentration (lactate, potassium, excitatory amino acids [EAA]) or decreased levels (glucose) after head injury have also been reported [60-64]. Belli et al. noted that elevated lactate/pyruvate (LP) ratios and glycerol levels preceded malignant increases in ICP, suggesting that biochemical failure could be detected early [65] (Figure 36.6).
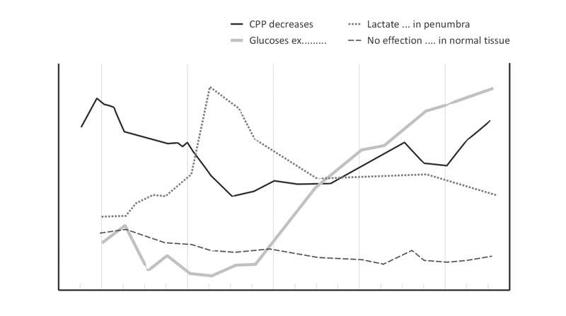
Figure 36.5. Effcects of lactate/pyruvate and glycerol levels on ICP.
Such cerebral substrates can be collected utilizing a fine coaxial probe (diameter, 0.62 mm) with a dialysis membrane (typically 20 kDa MW cut-off) placed directly into the brain. The probe is slowly perfused with a sterile mock extracellular fluid (ECF) solution, allowing continuous sampling of the parenchymal extracellular environment across the dialysis membrane [66]. High-performance liquid chromatography (HPLC) techniques in small aliquots of dialysate fluid (60 µl) allow for the identification and measurement of various molecules, including glucose, lactate, potassium, pyruvate, nitric oxide, and glutamate [60,61,67].
This tool was first utilized clinically in the 1980s by Meyerson et al. prior to lesioning for Parkinson’s disease [68] (Figure 36.6). As with most monitoring systems, microdialysis may be combined with other modalities such as ICP and PO2 probes [69]. Microdialysis has many limitations, however, one being that measurements are representative of relative concentrations of the extracellular molecules and so do not represent absolute concentrations, creating a problem when attempting to compare data between different machines and centres [70]. Other disadvantages are the lack of specificity of some results, sampling limitations of the microenvironment surrounding the catheter induced by cell edema, and changes in the ECF space [35,58,71,72], poor temporal resolution, and lengthy (1 hour) substrate analysis time. The use of microdialysis probes with a much higher molecular-weight cut-off (e.g., 100 kDa) allows protein fragments and small peptides such as cytokines (e.g., IL-1, IL-6, trophic factors, and amyloid precursor protein fragments) to be measured serially in the human brain following TBI and subarachnoid hemorrhage (SAH) [73]. Ongoing clinical investigations may address these drawbacks and eventually propel microdialysis into mainstream neurological ICU use.
Figure 36.6. Microdyalisis system.
36.7.4 Monitoring of Seizures and Spreading Depression: EEG/ECoG/CFM
Monitoring for epileptiform activity in the acute and subacute period post-TBI is an important adjunct to ICP, brain oximetry and CBF, as the incidence of subclincal/non-motor or partial seizures can be as high as 17-20% and chemical paralysis may mask overt motor seizures [35,74,75]. Electroencephalographic (EEG) data have also be used as an independent prognostic indicator and to institute therapeutic interventions before irreversible damage ensues [76,77]. The presence of epileptiform activity has also been linked to episodic increases in ICP and lactate/pyruvate ratios as measured by microdialysis [78]. Monitoring should continue until the ICP has normalized or cerebrovascular autoregulation has been re-established [78]. Additional benefits of obtaining EEG data post-TBI include testing the efficacy of new drugs without lengthy waiting to determine outcome measures. Vespa et al. studied 94 moderate to severe TBI patients and reported a 22% non-convulsive seizure rate. Classen et al. validated this study in 2004 and reported a 18% seizure rate in the same population.
Newer continuous EEG machines are smaller and portable, contain data reduction software, and real-time remote analysis, eliminating the need for a full-time technical assistant at the bedside [66]. The simplest form of EEG monitoring includes a non-invasive strip of 2-4 electrodes (e.g., Bispectral Index [BIS]) that analyzes the depth of sedation, reports on whether seizure activity is present, and can correlate with the GCS score [79]. A downside of this technique is that it cannot regionalize information. Larger units including 8-14 channel 10-12 electrode montages that are more regionally specific and can collect specific information such as alpha, delta, beta, and theta proportions of the raw EEG [76-78]. Several international studies are currently evaluating monitoring of cortical activity following decompressive craniectomy [80,81]. During the procedure, an electrocorticogram (ECoG) 6-electrode recording strip is placed on an area of the cortex near the parenchymal site of injury. Incorporated software provides data analysis and storage. The strip allows for bedside removal once the monitoring period is complete.
Cortical spreading depression (CSD), as previously described by Leao, refers to a wave of depressed cortical activity or amplitude that spreads slowly across the cerebral grey matter and can be diagnosed by the ECoG [82]. The depression is accompanied by a transient loss of ion homeostasis, changes in glucose and lactate, and an increase in CBF and oxygen followed by oligaemia. ECoG changes are similar to those recorded during peri-infarct depolarization (PID) which occur in the periphery of infarct regions [80,83-85]. PID and CSD differ from one another, however, in that PID contributes to irreversible ischemic damage, while it is unclear whether CSD can harbour long-term effects. It may precede certain events (e.g., seizures) and in some cases may even have a protective effect on the injured brain [81,86,87]. Differentiation between these two phenomena is critical.
CSD is not caused by one single mechanism. Animal studies suggest the influence of many different causative agents such as potassium and glutamate that flush out of the cells and into the ECF [88]. Although experimental work on this concept in animals spans 60 years, it has only recently been demonstrated that CSD post-TBI in humans is common, and it may even be more likely to occur in younger patients [80,89,90].
Fabricius et al. examined the incidence of co-occurrence of spreading depression and seizures following acute brain injury [81]: 54 patients were included in the study and each had a 6-electrode cranial strip placed below their craniotomy site; 19 of 54 patients exhibited cortical spreading depression, and in 8 of these 19 patients co-occurrence of seizures and CSD was documented. Three of these 19 patients had CSD that preceded a seizure, signifying the potential of CSD to trigger full seizures; however, this was not seen consistently. Only one of the seizures was observed clinically. More research into these two potentially co-occurring phenomena is necessary. Because CSD and PID events in animals can be attenuated with N-methyl-D-aspartic (NMDA) acid receptor blockade, monitoring these changes in humans may provide an important selection tool for those patients who could benefit from these therapies in the future [90].
36.7.5 Neuromonitoring-based Treatment Strategies
As mentioned earlier, the goals of monitoring and treatment are to maintain adequate cerebral perfusion, oxygenation, and metabolism, while limiting ICP elevation, desaturation, edema, and substrate perturbations. Specific values that may serve as a guide include maintenance of CPP between 60 and 70 mmHg, ICP <20 mmHg, SjvO2 >50 mmHg, and local tissue oxygenation >15-20 mmHg. Traditionally, treatment of elevated ICP has proceeded in a step-wise fashion, beginning with a low-risk and high-benefit profile before progressing to high-risk therapy such as barbiturate coma. Elevation of the head of the bed to 30-45 degrees, maintenance of normotension, normothermia, and normocarbia, prevention of hypoxia, and sedation and analgesia remain the initial steps in the management of most head-injured patients. These are first-tier treatments in severe TBI. Progression to higher tier therapies should be initiated when there is a rise in ICP, and substrate deficiencies in multi-modality monitoring require some unique and specific interventions, as discussed below.
Osmotic Diuretics
Mannitol, currently the osmotic diuretic of choice, is initially given as a bolus of 0.25-1 gm/kg. It reduces ICP within 15 minutes, and its effects typically persist for 3-4 hours. The therapeutic effects of mannitol include initial improvement in red blood cell rheology, blood flow and oxygen delivery to the brain, as well as delayed osmotic water withdrawal from the brain into the circulating blood volume [91]. There is also weak evidence that concomitant use of loop diuretics, such as furosemide, results in a greater and more sustained decrease in ICP when compared to mannitol use alone [92]. Clinical effects seem to peak when serum osmolarity is kept between 300 and 320 mOsm, and elevations beyond 320 mOsm can result in renal failure. Some authors advocate the use of hypertonic saline, but evidence does not support a superior effect in TBI.
CSF Drainage
The first step in managing a new rise in ICP is to rule out an evolving or delayed mass lesion by CT scan. Once this has occurred, and assuming a ventriculostomy is in place, drainage of 3-5 ml of CSF should be the initial therapeutic modality. In patients with intracranial hypertension, drainage of even a small amount of CSF may be very effective. An additional theoretical advantage of CSF drainage involves removal of potentially neurotoxic compounds from the CSF, such as glutamate, aspartate, and calcium. These compounds have been shown to be present after TBI, and facilitation of their removal may have a theoretical advantage.
Sedation and Neuromuscular Paralysis
An additional therapeutic option involves heavy sedation and/or neuromuscular paralysis. Agitation, anxiety, posturing, and resistance to mechanical ventilation can all contribute to elevations in ICP. Sedatives, such as midazolam or lorazapam, propofol coupled with narcotics such as morphine or fentanyl, and neuromuscular blocking agents such as vecuronium or rocuronium, are all commonly used agents. Specifics on dosing and benefit/side effect profiles are beyond the scope of this chapter. Routine neuromuscular paralysis of head-injured patients has been shown to increase pulmonary complications and prolong ICU stay however, and should therefore be reserved for those patients with persistent elevation in ICP [93].
Hyperventilation
Lundberg was the first to introduce hyperventilation as a form of treatment for intracranial hypertension in the 1950s, associating a significant decrease in ICP in patients with tumour or TBI when arterial PCO2 was reduced from 40 to 25 mmHg [94]. Subsequent animal models confirmed that hyperventilation leads to cerebral vasoconstriction, decreased CBF and blood volume, and a reduction in ICP. Research has shown that CBF in the pericontusional area is indeed close to the ischemic/infarction threshold (<18 ml/ gm/min) and that further reductions in blood flow may tip this balance towards brain ischemia. Routine hyperventilation to an arterial PCO2 of 20-25 mmHg was shown to worsen outcome in a randomized controlled trial [95]. It is now recommended that PCO2 be kept at or near 35 mmHg whenever possible. Aggressive hyperventilation should only be employed to treat acute changes and only for short periods of time; and SjvO2 or PTiO2 measurements should augment these treatments to ensure adequate oxygenation.
Decompressive Craniectomy
Although surgical indications will be addressed in subsequent chapters, decompressive craniectomy for treating refractory intracranial hypertension will be discussed briefly here. Decompressive surgical procedures remain controversial, but there is some evidence that they may reduce mortality rates. For example, Gower et al. reported a reduction in mortality from 80 to 40% in patients undergoing subtemporal decompression in the face of refractory intracranial hypertension. The “Guidelines for the Surgical Management of Traumatic Brain Injury” recommend the following: “Decompressive procedures, including subtemporal decompression, temporal lobectomy, and hemispheric decompressive craniectomy, are treatment options for patients with refractory intracranial hypertension and diffuse parenchymal injury with clinical and radiographic evidence of impending herniation”.
Two ongoing clinical trials, the Rescue-ICP (British Medical Research Council) and the DECRA (Australian Medical Research Council), should clarify the role of this treatment in the next few years.
Barbiturate Coma
Barbiturate use is considered the last line of treatment options for patients with refractory intracranial hypertension. Its use may be beneficial due to the action of different mechanisms, including free radical scavenging, lowering of cerebral metabolic rate, cerebral vasoconstriction in uninjured areas shunting blood to injured areas, calcium homeostasis, and lysosomal stabilization. Although routine use of barbiturates in unselected patients has not been shown to reduce morbidity or mortality after severe head injury, Eisenberg et al., in a randomized multicentre trial, showed a two-fold greater chance of controlling ICP in patients with refractory intracranial hypertension when barbiturate coma was induced [96].
Since barbiturates can cause profound hypotension, and neurologic exams cannot be performed during its use, patients with cardiovascular instability are not usually candidates. Pentobarbital is the most common agent, and systemic monitoring with Swan-Ganz catheters is recommended. Barbiturates can be useful in a select group of patients however, and those with sustained refractory intracranial hypertension likely have no other option.
Hypothermia
Studied and used intermittently for decades, hypothermia is attracting renewed interest. Therapeutic moderate hypothermia (32-35°C) has been shown to lower ICP and has been linked to improved outcome after TBI [97,98]. Hypothermia reduces CBF, metabolism, and inflammation, much akin to barbiturate use. At our institution, we treat most severe head injuries with 48-72 hours of moderate hypothermia, instituted as soon as possible after the injury.
36.8 Conclusions
Traumatic brain injury is common worldwide; treatment outcomes continue to improve through continued research and clinical advances. The trauma victim should be rapidly identified, stabilized and transferred to a trauma care facility where care should be smoothly transferred to an awaiting multidisciplinary team ready to combat all possible injuries. Rapid cardiovascular stabilization, radiographic diagnosis, and intervention should be the goal in every case. Treatment in the ICU should continue with the same vigilance and attention, as many outcomes continue to be determined by ICU management. Multi-modality neuro-monitoring continues to expand through information and randomized controlled trials, and new product developments and improvements. Most modalities on their own do not paint a clear portrait, but when combined the synergy of multi-modality monitoring has the potential for the sum to be greater than the parts. All of these products show great potential for application in appropriate contexts and interventions. Their wider acceptance may ultimately lead to better patient outcomes.
References
1. Finkelstein E, Corso P, Miller T (eds). The incidence and economic burden of injuries in the United States. New York (NY): Oxford University Press, 2006
2. Dawodu S. Traumatic Brain Injury (TBI)-Definition, Epidemiology, Pathophysiology. 2009 Available at: http://emedicine.medscape.com/article/326510-overview
3. Klauber MR, Marshall LF, Toole BM, et al. Cause of decline in head-injury mortality rate in San Diego County, California. J Neurosurg 1985; 62: 528-31
4. Stocchetti N, Furlan A, Volta F. Hypoxemia and arterial hypotension at the accident scene in head injury. J Trauma 1996; 40: 764-7
5. Mendelow AD, Teasdale G, Jennett B, et al. Risks of intracranial hematoma in head injured adults. Br Med J (Clin Res Ed) 1983; 287: 1173-6
6. Gennarelli TA. Graham DI. Neuropathology of the Head Injuries. Semin Clin Neuropsychiatry 1998; 3: 160-75
7. McIntosh T, Saatman K, Raghupathi R. Calcium and the pathogenesis of traumatic CNS injury: Cellular and molecular mechanisms. Neuroscientist 1997; 3: 169-75
8. Faden AI, Demediuk P, Panter SS, et al. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989; 244: 798-800
9. Katayama Y, Becker DP, Tamura T, et al. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg 1990; 73: 889-900
10. Unterberg A, Wahl M, Hammersen F, et al. Permeability and vasomotor response of cerebral vessels during exposure to arachidonic acid. Acta Neuropathol 1987; 73: 209-19
11. Raghupathi R, Welsh FA, Lowenstein DH, et al. Regional induction of c-fos and heat shock protein-72 mRNA following fluid-percussion brain injury in the rat. J Cereb Blood Flow Metab 1995; 15: 467-73
12. Whitfield C, Pickard JD. Expression of the immediate early genes c-Fos and c-Jun after head injury in man. Neurol Res 2000; 22: 138-44
13. Rink A, Fung KM, Trojanowski JQ, et al. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol 1995; 147: 1575-83
14. Clark RS, Kochanek PM, Chen M, et al. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. Faseb J 1999; 13: 813-21
15. Bredesen DE. Apoptosis: overview and signal transduction pathways. J Neurotrauma 2000; 17: 801-10
16. Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma 2000; 17: 811-29
17. Shohami E, Novikov M, Bass R, et al. Closed head injury triggers early production of TNF alpha and IL-6 by brain tissue. J Cereb Blood Flow Metab 1994; 14: 615-9
18. Miller J, Piper IR, Jones, Pathophysiology of head injury. Neurotrauma 1996: 61-69
19. Chesnut RM, Marshall SB, Piek J, et al. Early and late systemic hypotension as a frequent and fundamental source of cerebral ischemia following severe brain injury in the Traumatic Coma Data Bank. Acta Neurochir Suppl (Wien) 1993; 59: 121-5
20. Kety SS, Schmidt CF. The nitrous oxide method for the quantitative determination of cerebral blood flow in man: theory, procedure and normal values. J Clin Invest 1948; 27: 476-83
21. Gibbs E. Arterial and cerebral venous blood: Arterial-venous differences in man. J Biol Chem 1942; 144: 325-332
22. Siesjo BK. Cerebral circulation and metabolism. J Neurosurg 1984; 60: 883-908
23. Muizelaar JP, Marmarou A, DeSalles AA, et al. Cerebral blood flow and metabolism in severely head-injured children. Part 1: Relationship with GCS score, outcome, ICP, and PVI. J Neurosurg 1989; 71: 63-71
24. Obrist WD, Langfitt TW, Jaggi JL, et al. Cerebral blood flow and metabolism in comatose patients with acute head injury. Relationship to intracranial hypertension. J Neurosurg 1984; 61: 241-53
25. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980; 288(5789): 373-6
26. Bergsneider M, Hovda DA, Shalmon E, et al. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J Neurosurg 1997; 86: 241-51
27. Adams J, Graham D. The pathology of blunt head injury. Scientific Foundation of Neurology 1972: 488-491
28. Bouma G. Cerebral circulation after severe head injury: A clinical study, in University of Amsterdam.Amsterdam: University of Amsterdam, 1993
29. Jones TH, Morawetz RB, Crowell RM, et al. Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg 1981; 54: 773-82
30. Becker DP, Miller JD, Ward JD, et al. The outcome from severe head injury with early diagnosis and intensive management. J Neurosurg 1977; 47: 491-502
31. Marshall LF, Gautille T. Large and small “holes” in the brain: reversible or irreversible changes in head injury. Acta Neurochir Suppl (Wien) 1990; 51: 300-1
32. Miller JD. Physiology of trauma. Clin Neurosurg 1982; 29: 103-30
33. Chesnut RM. Treating raised intracranial pressure in head injury. Neurotrauma 1996: 445-69
34. Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: management protocol and clinical results. J Neurosurg 1995; 83: 949-62
35. Guidelines for the management of severe traumatic brain injury. J Neurotrauma 2007; 24 Suppl 1: S1-106
36. Czosnyka M, Czosnyka Z, Pickard JD. Laboratory testing of three intracranial pressure microtransducers: technical report. Neurosurgery 1996; 38: 219-24
37. Gupta AK, Bullock MR. Monitoring the injured brain in intensive care: present and future. Hosp Med 1998; 59: 704-13
38. Lindegaard KF, Nornes H, Bakke SJ, et al. Cerebral vasospasm diagnosis by means of angiography and blood velocity measurements. Acta Neurochir (Wien) 1989; 100: 12-24
39. McCormick PW, Stewart M, Goetting MG, et al. Noninvasive cerebral optical spectroscopy for monitoring cerebral oxygen delivery and hemodynamics. Crit Care Med 1991; 19: 89-97
40. Kirkpatrick J. Near-infrared spectroscopy use in patients with head injury. J Neurosurg 1995; 83: 963-70
41. Brazy JE. Cerebral oxygen monitoring with near infrared spectroscopy: clinical application to neonates. J Clin Monit 1991; 7: 325-34
42. Jobsis FF. Noninvasive, infrared monitoring of cerebral and myocardial oxygen sufficiency and circulatory parameters. Science 1977; 198: 1264-7
43. McCormick PW, Stewart M, Goetting MG, et al. Regional cerebrovascular oxygen saturation measured by optical spectroscopy in humans. Stroke 1991; 22: 596-602
44. Brown JI, Moulton RJ, Konasiewicz SJ, et al. Cerebral oxidative metabolism and evoked potential deterioration after severe brain injury: new evidence of early posttraumatic ischemia. Neurosurgery 1998; 42: 1057-63; discussion 1063-4
45. Germon TJ, Evans PD, Manara AR, et al. Sensitivity of near infrared spectroscopy to cerebral and extra-cerebral oxygenation changes is determined by emitter-detector separation. J Clin Monit Comput 1998; 14: 353-60
46. Kirkpatrick PJ, Smielewski P, Whitfield PC, et al. An observational study of near-infrared spectroscopy during carotid endarterectomy. J Neurosurg 1995; 82: 756-63
47. Carter LP, Weinand ME, Oommen KJ. Cerebral blood flow (CBF) monitoring in intensive care by thermal diffusion. Acta Neurochir Suppl (Wien) 1993; 59: 43-6
48. Sheinberg M, Kanter MJ, Robertson CS, et al. Continuous monitoring of jugular venous oxygen saturation in head-injured patients. J Neurosurg 1992; 76: 212-7
49. Robertson C. Desaturation episodes after severe head injury: influence on outcome. Acta Neurochir Suppl (Wien) 1993; 59: 98-101
50. Gopinath SP, Robertson CS, Contant CF, et al. Jugular venous desaturation and outcome after head injury. J Neurol Neurosurg Psychiatry 1994; 57: 717-23
51. Siggaard-Andersen O, Fogh-Andersen N, Gøthgen IH, et al. Oxygen status of arterial and mixed venous blood. Crit Care Med 1995; 23: 1284-93
52. Sarrafzadeh AS, Kiening KL, Unterberg AW. Neuromonitoring: brain oxygenation and microdialysis. Curr Neurol Neurosci Rep 2003; 3: 517-23
53. Kiening KL, Unterberg AW, Bardt TF, et al. Monitoring of cerebral oxygenation in patients with severe head injuries: brain tissue PO2 versus jugular vein oxygen saturation. J Neurosurg 1996; 85: 751-7
54. Lang EW, Mulvey JM, Mudaliar Y, et al. Direct cerebral oxygenation monitoring–a systematic review of recent publications. Neurosurg Rev 2007; 30: 99-106; discussion 106-7
55. Mazzeo AT, Bullock R. Monitoring brain tissue oxymetry: will it change management of critically ill neurologic patients? J Neurol Sci 2007; 261: 1-9
56. Scheufler K. Tissue oxygenation and capacity to deliver O2 do the two go together? Transfus Apheresis Sci 2004; 31: 45-54
57. van den Brink WA, van Santbrink H, Steyerberg EW, et al. Brain oxygen tension in severe head injury. Neurosurgery 2000; 46: 868-76; discussion 76-8
58. Sarrafzadeh AS, Kiening KL, Bardt TF, et al. Cerebral oxygenation in contusioned vs. nonlesioned brain tissue: monitoring of PtiO2 with Licox and Paratrend. Acta Neurochir Suppl 1998; 71: 186-9
59. Metz C, Holzschuh M, Bein T, et al. Moderate hypothermia in patients with severe head injury: cerebral and extracerebral effects. J Neurosurgery 1996; 85: 533-41
60. Reinert M, Khaldi A, Zauner A, et al. High level of extracellular potassium and its correlates after severe head injury: relationship to high intracranial pressure. J Neurosurg 2000; 93: 800-7
61. Bullock MR, et al., New techniques for multi-modality monitoring of the injured brain: opportunities for therapy? 1997; 9(11): 1295-300
62. Bullock R, Zauner A, Myseros JS, et al. Evidence for prolonged release of excitatory amino acids in severe human head trauma. Relationship to clinical events. Ann N Y Acad Sci 1995; 765: 290-7; discussion 8
63. Persson L, Hillered L. Chemical monitoring of neurosurgical intensive care patients using intracerebral microdialysis. J Neurosurg 1992; 76: 72-80
64. Hutchinson PJ, al-Rawi PG, O’Connell MT, et al. On-line monitoring of substrate delivery and brain metabolism in head injury. Acta neurochirurgica 2000; 76: 431-5
65. Belli A, Sen J, Petzold A, Russo S, Kitchen N, Smith M. Metabolic failure precedes intracranial pressure rises in traumatic brain injury: a microdialysis study. Acta Neurochir (Wien) 2008; 150: 461-9; discussion 70
66. Reilly, Bullock R. Head injury: pathophysiology and management. 2nd ed. London-New York: Hodder Arnold, 2005; distributed in the U.S.A. by Oxford University Press. x, 501 p
67. Khaldi A, Zauner A, Reinert M, et al. Measurement of nitric oxide and brain tissue oxygen tension in patients after severe subarachnoid hemorrhage. Neurosurgery 2001; 49: 33-8; discussion 38-40
68. Meyerson BA, Linderoth B, Karlsson H, et al. Microdialysis in the human brain: extracellular measurements in the thalamus of parkinsonian patients. Life Sci 1990; 46: 301-8
69. Sarrafzadeh AS, Sakowitz OW, Callsen TA, et al. Bedside microdialysis for early detection of cerebral hypoxia in traumatic brain injury. Neurosurg Focus 2000; 9: e2
70. Johnston AJ, Gupta AK. Advanced monitoring in the neurology intensive care unit: microdialysis. Curr Opin Crit Care 2002; 8: 121-7
71. Bullock MR, Doppenberg E, Zauner A, et al. New Techniques for Multi-Modality Monitoring of the Injured Brain: Opportunities for Therapy? 1997; 9: 1295-300
72. Meixensberger J, Kunze E, Barcsay E, et al. Clinical cerebral microdialysis: brain metabolism and brain tissue oxygenation after acute brain injury. Neurological research 2001; 23: 801-6
73. Hillman J, Aneman O, Anderson C, et al. A microdialysis technique for routine measurement of macromolecules in the injured human brain. Neurosurgery 2005; 56: 1264-8; discussion 8-70;
74. Vespa PM. Continuous EEG monitoring for the detection of seizures in traumatic brain injury, infarction, and intracerebral hemorrhage: “to detect and protect”. J Clin Neurophysiol 2005; 22: 99-106
75. Vespa PM, Nuwer MR, Nenov V, et al. Increased incidence and impact of nonconvulsive and convulsive seizures after traumatic brain injury as detected by continuous electroencephalographic monitoring. J Neurosurg 1999; 91: 750-60
76. Hebb MO, McArthur DL, Alger J, et al. Impaired percent alpha variability on continuous electroencephalography is associated with thalamic injury and predicts poor long-term outcome after human traumatic brain injury. J Neurotrauma 2007; 24: 579-90
77. Vespa PM, Boscardin WJ, Hovda DA, et al. Early and persistent impaired percent alpha variability on continuous electroencephalography monitoring as predictive of poor outcome after traumatic brain injury. J Neurosurg 2002; 97: 84-92
78. Vespa PM, Miller C, McArthur D, et al. Nonconvulsive electrographic seizures after traumatic brain injury result in a delayed, prolonged increase in intracranial pressure and metabolic crisis. Crit Care Med 2007
79. Paul D, Rao G. Correlation of bispectral index with Glasgow Coma Score in mild and moderate head injury. J Clin Monit Comput 2006; 20: 399-404
80. Strong AJ, Fabricius M, Boutelle MG, et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 2002; 33: 2738-43
81. Fabricius M, Fuhr S, Willumsen L, et al. Co-occurence of spreading depression and seizures after acute brain injury. Clin Neurophysiol 2008 119: 1973-84
82. Leao A. Spreading depression of activity in cerebral cortex. J Neurophysiol 1944; 7: 359-90
83. Hopwood SE, Parkin MC, Bezzina EL, et al. Transient changes in cortical glucose and lactate levels associated with peri-infarct depolarisations, studied with rapid-sampling microdialysis. J Cereb Blood Flow Metab 2005; 25: 391-401
84. Lauritzen M, Jorgensen MB, Diemer NH, et al. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann Neurol 1982; 12: 469-74
85. Hossman K. Periinfarct depolarizations. Cerebrovasc Brain Metab Rev 1996; 8: 195-208
86. Kraig RP, Nicholson C. Extracellular ionic variations during spreading depression. Neuroscience 1978; 3: 1045-59
87. Busch E, Gyngell M, Elis M, Hoehn Berlage M, Hossman K. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab 1996; 16: 1090-9
88. Seelig JM, Becker DP, Miller JD, et al. Traumatic acute subdural hematoma: major mortality reduction in comatose patients treated within four hours. N Engl J Med 1981; 304: 1511-8
89. Martins-Ferreira H, Nedergaard M, Nicholson C. Perspectives on spreading depression. Brain Res Brain Res Rev 2000; 32: 215-34
90. Fabricius M, Fuhr S, Bhatia R, et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 2006; 129: 778-90
91. Muizelaar JP, Wei EP, Kontos HA, et al. Mannitol causes compensatory cerebral vasoconstriction and vasodilation in response to blood viscosity changes. J Neurosurg 1983; 59: 822-8
92. Pollay M, Fullenwider C, Roberts PA, et al. Effect of mannitol and furosemide on blood-brain osmotic gradient and intracranial pressure. J Neurosurg 1983; 59: 945-50
93. Hsiang JK, Chesnut RM, Crisp CB, et al. Early, routine paralysis for intracranial pressure control in severe head injury: is it necessary? Crit Care Med 1994; 22: 1471-6
94. Lundberg N, Kjallquist A, Bien C. Reduction of increased intracranial pressure by hyperventilation. A therapeutic aid in neurological surgery. Acta Psychiatr Scand Suppl 1959; 34: 1-64
95. Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg 1991; 75: 731-9
96. Eisenberg HM, Frankowski RF, Contant CF, et al. High-dose barbiturate control of elevated intracranial pressure in patients with severe head injury. J Neurosurg 1988; 69: 15-23
97. Shiozaki T, Sugimoto H, Taneda M, et al. Effect of mild hypothermia on uncontrollable intracranial hypertension after severe head injury. J Neurosurg 1993; 79: 363-8
98. Marion DW, Penrod LE, Kelsey SF, et al. Treatment of traumatic brain injury with moderate hypothermia. N Engl J Med 1997; 336: 540-6
Related posts:
 Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
 Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation
Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation
 Extracranial Atherosclerotic Carotid Artery Disease
Extracranial Atherosclerotic Carotid Artery Disease
 Acute Paraplegias and Quadriplegias of Non-traumatic Cause
Acute Paraplegias and Quadriplegias of Non-traumatic Cause
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


