CHAPTER 148 Skull Tumors
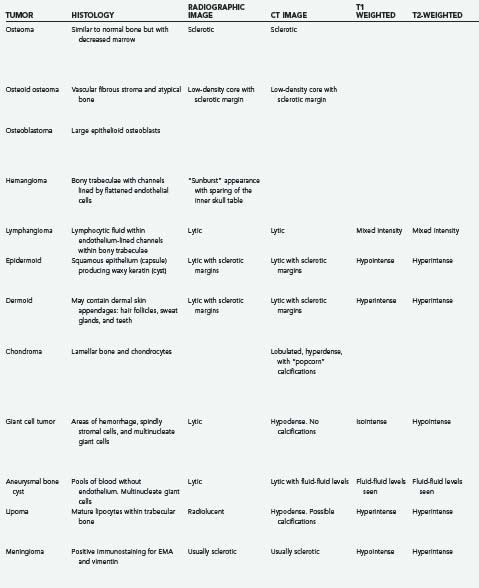
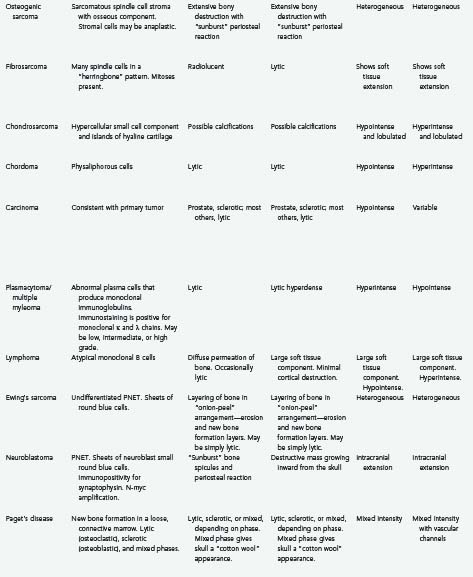
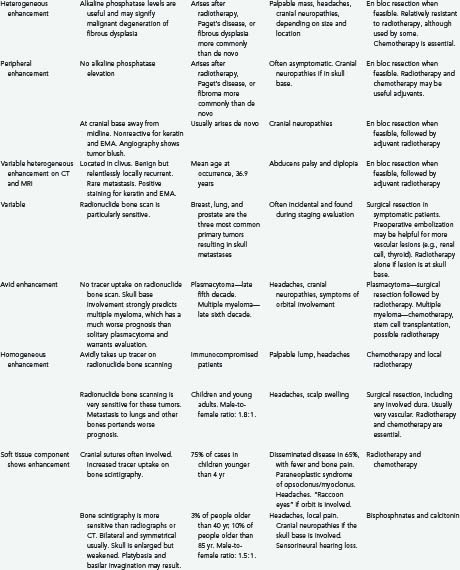
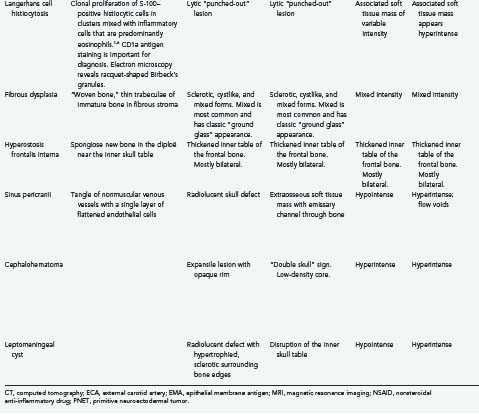
Tumors of the skull make up a small but significant portion of neurosurgical practice. These lesions may be primary neoplastic, secondary neoplastic, or non-neoplastic. Their characteristics and treatments are summarized in Table 148-1.1–6

Primary Skull Neoplasms
Benign
Osteoma/Osteoid Osteoma/Osteoblastoma
Osteomas are the most common primary skull lesions.7 These benign, slow-growing tumors have a predilection for the craniofacial bones and paranasal sinuses and consist of abnormally dense but otherwise normal bone.7–11 Osteomas of the calvaria typically arise from the external table of the skull but may also arise from the inner table.11 Their most common location is the frontal sinus, followed by the ethmoid sinus, which together account for 75% of cases.8,11 Osteomas of the maxillary sinus and sphenoid sinus have been reported as well.8,11 These lesions are often asymptomatic but may cause local swelling and tenderness, facial pain, rhinorrhea, and sinusitis.8,12 Additionally, osteomas tend to expand bone, which may lead to displacement of the frontal lobes and orbital contents.10 Subsequent erosion of the dura and arachnoid may cause cerebrospinal fluid (CSF) rhinorrhea with resultant meningitis or brain abscess.10 If sinus mucosa herniates through the eroded bone into the intracranial space, a mucocele may develop.10 En bloc surgical resection of osteomas is often curative and is indicated even for asymptomatic lesions if they grow rapidly or involve the orbit.7
Histologically, osteomas generally consist of mature bone similar to normal bone, albeit with decreased marrow.9 There are three histopathologic types of osteoma: compact, cancellous, and fibrous. The compact (or “ivory”) type tends to occur over the outer table of the skull, whereas the cancellous and fibrous types tend to involve the inner table or, rarely, the diploë.2
Osteomas produce osteoblastic changes that may appear as zones of hyperostosis on radiography or computed tomography (CT).8,11 They typically appear as a smoothly homogeneous, sharply demarcated sclerotic mass extending outward from the bone without exerting any mass effect on adjacent soft tissue.8,11
Multiple osteomas may be associated with Gardner’s syndrome—an autosomal dominant disease characterized by the triad of osteomatosis, colonic polyposis, and soft tissue tumors.11,13 The lesions may affect the skull proper, as well as the mandible, facial bones, and paranasal sinuses.13 Osteomas have also been reported in association with tuberous sclerosis.8,11 Evaluation of patients with paranasal osteomas should thus include colonoscopy and DNA testing.12
Initially described by Jaffe in 1953 and first noted in the skull by Prabhakar and associates in 1972, osteoid osteomas are bone-forming neoplasms characterized by the production of osteoid or mature bone by tumor cells.8,9,14 These tumors are associated with swelling and local tenderness, particularly at night, which respond dramatically to nonsteroidal anti-inflammatory drugs (NSAIDs).8,14–17 This pain is often quite severe relative to the small size of the lesion.15 Osteoid osteoma is more common in adolescents and young adults and is relatively uncommon after 30 years of age.16,17 There is a distinct male predilection (1.7 : 1).16,17 The lesion typically develops slowly and may actually cause symptoms before being discernible on plain radiographs.16
Histologically, osteoid osteoma is characterized by trabeculae of atypical bone within a highly vascular fibrous stroma.1,14,16
Radiologically, this lesion is manifested on plain radiographs and CT by a low-density central nidus surrounded by a high-density sclerotic margin.1,14,15,18 Nuclear medicine bone scanning may be helpful in equivocal cases because the nidus of an osteoid osteoma lesion avidly “lights up” with tracer (less at the sclerotic margin).9 More recently, at least one report has suggested that magnetic resonance imaging (MRI) with gadolinium contrast enhancement demonstrates nidus conspicuity equal or superior to that seen with thin-section CT.19
With symptomatic or cosmetically deforming lesions, en bloc resection appears to be the treatment favored from an oncologic standpoint, although the risk of recurrence after simple curettage is low.15 Spontaneous regression of osteoid osteoblastomas has been reported.17
Histologically, osteoblastomas are closely related to osteoid osteomas and have been referred to as giant osteoid osteomas.9 Osteoblastomas share many of the same histologic features as osteoid osteomas, but they are greater than 2 cm in maximal diameter and are less responsive to NSAIDs for pain relief.20 Some histologic distinctions may include increased vascularity and a less organized pattern of osteoid and reticular bone in osteoblastomas.9 Some believe that although osteoid osteomas tend toward spontaneous regression,17 osteoblastomas may rarely undergo malignant transformation to osteosarcoma.9,20 These more aggressive osteoblastomas may be characterized histologically by “epithelioid” osteoblasts that are twice the size of normal osteoblasts.9 As with osteoid osteoma, en bloc surgical excision of osteoblastomas usually results in pain relief, and recurrence is rare.20 Treatment by partial resection or curettage is associated with a 10% tumor recurrence rate.15
Hemangioma/Lymphangioma
Although hemangiomas of bone are not uncommon, calvarial hemangiomas are relatively rare.21,22 These cranial vascular lesions are found more commonly in women than men (3 : 1 ratio)11,21–24 and have a predilection for the frontal and parietal areas.11,21,23,25 Hemangiomas vary in size from microscopic to massive and may be classified as either capillary or cavernous, depending on their histologic appearance.26 In 15% of cases, the lesions are multiple.21,24 Skull hemangiomas are often small and asymptomatic but may evolve into a visible and palpable area of tenderness and swelling.22,23 The scalp overlying the lesion is typically freely mobile.22 Hemangiomas in less common locations such as the base of the skull or orbit may cause cranial neuropathies or proptosis.21,25
Histologically, cavernous hemangiomas are unencapsulated sinusoidal channels lined by flattened endothelial cells within bony trabeculae. Capillary hemangiomas are similar but contain more capillary-sized vessel loops; frequently, a mixture of both histologic forms is present. The bony trabeculae are thought to be caused by osteoclastic remodeling in response to stress from the enlarging vascular malformation.1,21,25
Hemangiomas are always benign and may remain static for long periods.21,23,25 When they do grow, one of two growth patterns is typically exhibited: a “sessile” pattern involving extension along the diploë or a “globular” pattern with expansion of the skull and eventual extension into the surrounding soft tissues.5 The sessile type is more common.11 Association of skull hemangiomas with hemangiomas elsewhere in the body (including other bones), such as in the liver, kidney, spleen, and adrenal gland, has been reported, although true metastases have not.21
Calvarial hemangiomas tend to involve the outer table of the skull and the diploë, with relative sparing of the inner table of the skull.11,22 The classic appearance of a hemangioma on radiographs is a “sunburst” or “honeycomb” pattern.11,21,25 There is generally no surrounding sclerosis, and in fact, there may be a halo of decalcification.21,25 This pattern can perhaps best be appreciated on CT bone windows.24,25 Enhancement with contrast material is intense on both CT and MRI. Angiography may demonstrate feeding of the hemangioma by branches of the external carotid artery (ECA) and often by the superficial temporal artery or the middle meningeal artery,11,21,24 and it may suggest a pathway for preoperative embolization.22,25 Radionuclide bone scanning is of little use in the assessment of these lesions.5
En bloc surgical excision with establishment of normal bony margins (Fig. 148-1), when indicated, typically results in cure.21,23,25 Simple curettage may increase the chance of leaving residual disease and tumor recurrence.22 Some have advocated radiation treatment for residual tumor or for skull base lesions that are difficult to access surgically.20 Treatment of a symptomatic cavernous hemangioma of the occipital condyle with methacrylate embolization has also been reported.27
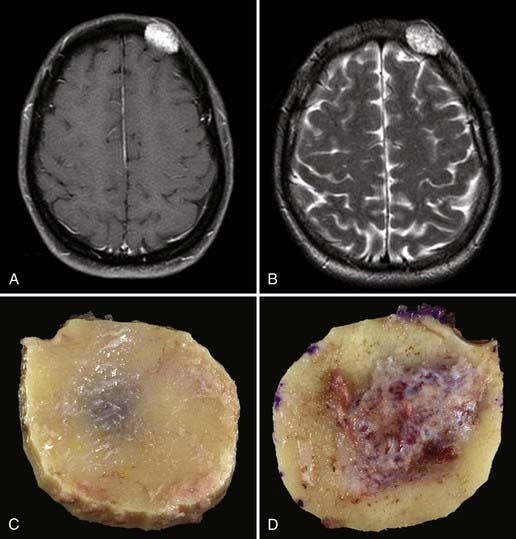
Lymphangioma is a congenital soft tissue tumor most commonly found in the neck; osseous involvement is rare.1,28,29 The first lymphangioma of bone was reported in 1947; the first solitary calvarial lesion without other visceral or skeletal involvement was noted in 1974.28,30 Skull base involvement is even more uncommon and has been reported to cause CSF rhinorrhea and cranial neuropathy.28,31 Lymphangioma is more common in the first 2 decades of life and is found with equal frequency in men and women.28,29,32 The most common finding is a palpable, possibly painful mass. Spontaneous bleeding into the mass has been described.29,32 One report described two particularly severe pediatric cases of lymphangiomatosis leading to massive osteolysis of the skull.33
Histologically, this benign lesion is very similar to hemangioma.28,30 Typically, the tumor is composed of large spaces lined with endothelium and filled with proteinaceous lymphocytic fluid within a fibrous connective tissue stroma. Bony trabeculae may also be present.1,29 The proportion of lymphocytic fluid to fibrous stroma may vary considerably.32 There are three histologic subtypes: cystic (cystic hygroma), capillary, and cavernous.28,29
As seen on plain radiographs, the lesions are lytic with a well-demarcated, nonsclerotic margin.29,30 Multiple lesions are not uncommon.29 The variable histologic makeup of lymphangioma causes the CT appearance to be similarly variable,32 and with the administration of contrast material, enhancement is weak if present at all.32 Lymphangiomas have a characteristic mixed-intensity, “bubbly” appearance on MRI sequences.32 Radionuclide bone scanning may be a useful diagnostic adjunct for these lesions.30 En bloc surgical resection is the treatment of choice for symptomatic or progressive lesions.28,29
Embryonic: Epidermoid and Dermoid
Epidermoid cysts and dermoid cysts together represent the epithelial inclusion cysts or “pearly tumors.”34,35 Both are thought to be formed in the third to fifth week in utero as a result of faulty separation of the ectoderm and neuroectoderm.36,37 Epidermoids are somewhat more common, are usually laterally situated, and are often found in adults.35,38 Dermoids are more commonly found in the midline and may be associated with congenital malformations.35,38–40
The first diploic epidermoid was described by Cushing in 1922. These cysts are usually congenital lesions caused by ectodermal rests within the skull diploë.38,40–43 Epidermoids have also been reported to be caused by traumatic induction of epidermal rests.40,44 Epidermoids are benign, slow-growing lesions, but they can undergo malignant change.40,45 They are typically asymptomatic, and a palpable painless lump is frequently the first sign.11,38,40,42 Giant intradiploic cysts can, however, cause elevated intracranial pressure and headaches.38,44,46 Rarely, intracranial extension occurs and results in seizures or venous sinus obstruction.11,38,40,42,47 Intracranial rupture plus hemorrhage of diploic epidermoids has also been reported after head trauma.40
Epidermoid cysts are usually well circumscribed, with a smooth, irregularly nodular capsule (Fig. 148-2).35 The lining of epidermoids is keratin-producing squamous epithelium.34,48 The cyst contains soft, white, waxy, grumous flaky keratin; leakage of the cyst contents into CSF may produce a severe chemical meningeal response known as Mollaret’s meningitis.34,35 Progressive exfoliation of the cyst wall toward the interior of the cyst causes a linear growth rate and gives its contents a lamellar appearance.11,35,38,48 The linear growth rate of the cyst is more similar to that of normal skin than to the exponential growth of most neoplasms.35
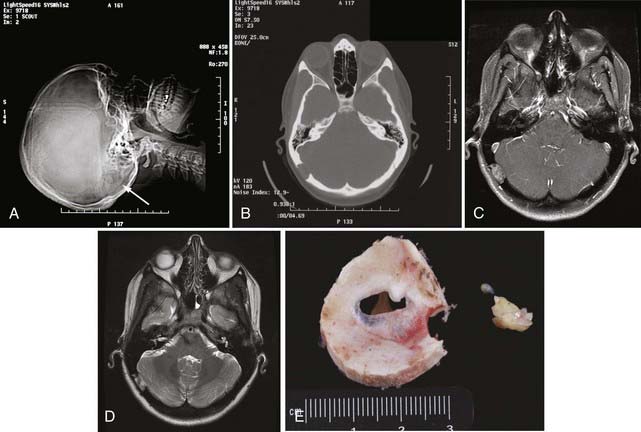
The radiographic appearance of epidermoids is that of a lytic lesion with sclerotic margins and diploic expansion.11,38,42,43,48 CT and MRI generally demonstrate a nonenhancing lesion with imaging characteristics similar to CSF, although they may be of mixed intensity on all imaging sequences and the capsule may show enhancement. Cholesterol within the cyst may give it a hypodense appearance on CT.11,38,43 CT is particularly useful for assessment of diploic expansion and for detecting destruction of the inner and outer skull tables, as well as any intracranial extension.38,41,44,48
Dermoid cysts are similar to epidermoids, although they are typically found in the midline, are more common in children and women, and may be associated with congenital malformations.35,38,39 They commonly occur at the anterior fontanelle and occipital squama.35,36,39,49,50
Dermoids grow by both desquamation and glandular secretion.51 As a result, the cyst typically contains thick foul-smelling yellow material in which hair may be entangled.35 Unlike epidermoids, the lining of dermoids contains dermal skin appendages: hair follicles, sweat glands, and, rarely, teeth.34,48
Similar to epidermoids, dermoids are often asymptomatic; their progressive outward expansion may lead to a palpable mass, and inward growth may compress the brain.39
Dermoids have CT and MRI characteristics similar to those of fat; they generally do not show contrast enhancement with either modality.51 Plain radiographs demonstrate an osteolytic region occasionally surrounded by a sclerotic border.49 CT shows a round, well-demarcated, hypodense lesion.51
Intracranial extension and involvement of the venous sinuses may be significant, and a preoperative magnetic resonance venogram is essential for determining this relationship.36,49,50 Dermoid tumors in the occipital region may also be associated with a dermal sinus tract and a consequent risk for meningitis or intracranial abscess.36,50
The treatment of choice for both types of inclusion tumor is complete surgical resection, which results in cure.38,40,42,44 A margin of normal bone is often resected to avoid disrupting the cyst capsule.41 Ideally, the tumor should be dissected away from the dura, although in some cases the dura must be resected as well.41 The risks related to incomplete surgical resection include recurrence, progression to squamous cell carcinoma, infection, and aseptic meningitis.35,36,39,41,44,45,52 The prognosis in patients whose tumors undergo malignant transformation is poor.41,44
Chondroma (Osteochondroma)
Chondromas (osteochondromas) are slow-growing benign tumors of cartilage. They are the most common benign skeletal lesion and are thought to arise from ectopic hyaline cartilaginous rests trapped within suture lines.1,53–57 In the skull, these lesions are so rare that Cushing found only 3 in his series of 2033 cases.58 Chondromas are well-demarcated tumors that are generally manifested as a smooth round mucosa-covered mass.1,58,59 Chondromas are most commonly found at the skull base (usually anterior to the pons), particularly in the sphenoid bone, or bordering the foramen lacerum,10,26,34,57,59,60 although convexity chondromas have been reported.10,57,60 Symptoms typically develop slowly and may include decreased visual acuity, ophthalmoplegia, tinnitus, dizziness, headaches, or facial pain.58,59 They may occasionally be associated with other chondromas in the skeleton, as in Ollier’s disease, or with multiple hemangiomas (Maffucci’s syndrome).56,60
Histologically, chondromas are composed of normal-appearing chondrocytes without pleomorphism or mitotic activity. Dense spicules of lamellar bone interspersed with marrow and fat cells are capped by cartilage of varying thickness.1,11,53,54,59 Generally, these tumors are confined by a capsule and do not invade the brain.10 Rare malignant degeneration of chondroma to chondrosarcoma has been documented.26,58
On CT, osteochondromas appear as well-demarcated, off-midline, lobulated, contrast-enhancing, dense masses contiguous with the underlying bone and marked by “popcorn” calcifications.53,54,59 Chondromas show avid contrast enhancement on MRI as well.59 Additionally, MRI is helpful for appreciating the thin cartilaginous cap overlying the dense osseous core.53,54 CT helps determine the extent of bone erosion.59
Complete resection, including the cartilaginous capsule, is the treatment of choice and is believed to be curative, although it is often difficult to achieve.54,56,59,60
Giant Cell Tumor
Giant cell tumors of bone are benign, locally aggressive lesions characterized by well-vascularized tissue infiltrated with multinucleate giant cells.39,61–66 Less than 2% of these lesions are found in the skull10,39,61,62; when present, they typically originate from the skull base, particularly the sphenoid or temporal bone.10,61,62,66 They usually appear in the second through fourth decades of life and cause destruction of the sphenoid bone and sella turcica.10,39,62,63 Initial symptoms may include headache, trigeminal paresthesias, ophthalmoparesis, visual loss, endocrinopathy, and changes in mental status.62,66 Lesions in the temporal bones may cause conductive hearing loss or facial weakness.61,62,66,67 Some giant cell tumors may arise in the skull secondary to Paget’s disease.39,64,68
Grossly, giant cell tumors are composed of cystic and hemorrhagic areas. Histologically, they are characterized by areas of hemorrhage, many spindly stromal cells, and numerous multinucleate giant cells.61,63,64 Molecular genetic analysis of laser microdissections suggest that the stromal cells are the true neoplastic component. The giant cells are a secondary inflammatory response rather than the primary element that the name implies. The extensive hemorrhagic areas may make it difficult to differentiate this lesion from an aneurysmal bone cyst.64,66 Giant cell tumors have a low risk for metastasis but are often locally aggressive, with a high risk for recurrence.62,64,67 However, there is little correlation between any of the histologic features of this lesion and its biologic behavior.66,67 A malignant variant of this tumor (osteoclastoma) was reported in one patient with Paget’s disease.10 Rare cases of intradural and intraparenchymal invasion have also been reported.61,66
Radiologically, these tumors are well-demarcated, lytic lesions without a sclerotic border or periosteal reaction.61,62,64,65 CT typically shows a homogeneously hypodense mass with avid enhancement and no calcifications.61,65 On MRI, the mass appears isointense on T1-weighted sequences and hypointense on both T2- and diffusion-weighted images.62,65 The presence of hemosiderin probably accounts for the hypointensity on T2 images.65
Gross total resection of giant cell tumors is ideal but not always feasible.61,62,66 The recurrence rate and prognosis associated with these lesions both correlate with the extent of resection.39,62,66 Simple curettage has been reported to have a tumor recurrence rate as high as 70%, whereas gross total resection has a recurrence rate of just 7%.63,64 En bloc removal with margins is best, but its feasibility is often limited by anatomic constraints. The choice of reoperation or radiotherapy for partially resected giant cell tumors is a matter of controversy61,66 in that there is the risk for malignant transformation if residual tumor is present and a risk for induction of sarcoma with irradiation.61,66 Good results with radiation treatment after tumor recurrence have been reported.66,67,69 Additionally, some authors have reported the use of chemotherapy for recurrent or metastatic tumors, or both, although this protocol remains controversial.62,67 The sarcoma regimen of methotrexate, doxorubicin (Adriamycin), and cyclophosphamide is most commonly used but is generally reserved for particularly aggressive tumors that have recurred despite multiple resections and radiation treatments.67
Aneurysmal Bone Cyst
Aneurysmal bone cysts are benign, non-neoplastic, expansile lesions of unknown cause that can occur anywhere in the body, including the skull in rare cases.1,39,70–72 Approximately 30% of cases are associated with antecedent trauma or with other lesions such as fibrous dysplasia and osteoblastoma. Recognition of aneurysmal bone cysts as a distinct clinical entity was thus initially met with some resistance.1,70,73–76 Lichtenstein hypothesized that the cysts are only secondarily associated with trauma and are more likely to be caused by a circulatory disturbance resulting in venous hypertension and venous pooling within the bone.74 These solitary, expansile, erosive lesions are more commonly found in young persons and are rare after the third decade of life.11,39,70,73,75 There is a 2 : 1 female-to-male preponderance.71,73 Aneurysmal bone cysts cause symptoms by local compression or cosmetic deformity71,75; a typical finding is local swelling and tenderness of a few months’ duration.39,73,75 Lesions at the skull base may cause ptosis, exophthalmos, visual loss, hearing loss, and facial weakness.71,75
Grossly, aneurysmal bone cysts are distinguished by vascular channels that give them a sponge-like appearance and account for brisk hemorrhage during surgical resection. Soft tissue and bone spurs may septate the mass, but the overall appearance is osteolytic.39,71 Histologic examination reveals communicating pools of venous blood without endothelium in a thin matrix of fibro-osseous strands along with frequent multinucleate giant cells. The appearance may often be similar to that of giant cell tumors, and distinguishing between the two entities can be difficult.1,39,64,66,70,71,73,75
On imaging studies, the finding of a lytic, loculated lesion with fluid-fluid levels caused by layering of blood products within internal cavities is highly suggestive of an aneurysmal bone cyst. MRI is superior to CT in visualizing these fluid-fluid levels.11,70,71,73,75 The lesion typically begins in the diploë, and expansion or “blowout” of the inner and outer cortices may be appreciated on plain radiographs or CT.70,71,73,75 Blowout of the inner skull table confirms the typical intracranial involvement of these lesions.71 The solid portion of the lesion may show contrast enhancement.70,71,76
The treatment of choice for aneurysmal bone cysts is gross total resection, which is curative when feasible.39,70,71,73,75 Preoperative embolization has been reported to be a helpful presurgical adjunct.70 The most important factor affecting tumor recurrence is the extent of resection; partial resection or curettage has been associated with recurrence rates as high as 71%.39,70,71,75 Other factors influencing recurrence include the age of the patient, the size of the lesion, and the presence of mitosis in the lesion.71 Because these lesions are benign and non-neoplastic, radiotherapy and chemotherapy are not indicated.70,71 Radiotherapy was used in the past but has fallen out of favor because of the risk of inducing sarcomatous change.71 Chartrand-Lefebvre and coauthors reported good results with sclerotherapy for a greater sphenoid wing lesion in one patient.77 Rare spontaneous regression of aneurysmal bone cysts of the skull has been reported.72
Lipoma
Intraosseous lipomas are rare lesions that represent 0.08% of all bone tumors; fewer than 20 cases have been reported in the world literature.78–84 They are generally benign, solitary, well-circumscribed, mobile, slow-growing, asymptomatic masses.1,81,83,84 The cause of lipomas arising in the skull is controversial.82 Some believe that the potential for intraosseous lipomas resides in the presence of fat cells in all skeletal bone marrow.35,81,83 Alternatively, intraosseous lipomas could arise from maldifferentiation of mesenchymal stem cells covering the maturing brain.83 Some have characterized intraosseous lipomas as hamartomas rather than true tumors.78
Histologically, skull lipomas consist of a proliferation of mature lipocytes within normal trabecular bone. A fibrous capsule may be present.78,80,83 At low magnification, lipomas resemble normal adipose tissue with lobulation.26,83
Radiologically, the lesions have the imaging characteristics of normal lipomatous tissue on CT and MRI, and this may obviate the need for biopsy.81,83 On plain radiographs, a well-demarcated, radiolucent lesion is noted, with occasional expansion of the diploë.81,82 CT demonstrates a hypodense expansile mass in the diploë separating the inner and outer tables, with or without periosteal reaction. Calcifications may be interspersed.78,80,81,83,84 Lipomas typically appear hyperintense on T1 sequences and isointense to hyperintense on T2 and fluid-attenuated inversion recovery (FLAIR) sequences.78,85
Skull lipomas are generally asymptomatic incidental findings that do not require treatment.78,81 One case has been reported of sphenoid bone involvement by an intraosseous lipoma with intrasellar and suprasellar extension that caused visual disturbance and hypopituitarism.86 In asymptomatic patients, observation with serial imaging studies is the standard of treatment78,81 because the risk for malignant degeneration is thought to be very low.81 Resection may be carried out for correction of cosmetic deformity or for compression-related symptoms.80,82,84,86 The recurrence rate after resection of these benign slow-growing tumors is as yet unknown.83
Meningioma
Intraosseous (intradiploic) meningiomas are uncommon. True primary intraosseous meningiomas do not involve the inner or outer tables of the skull or the dura.87 As meningiomas with no dural attachment, these tumors may be categorized as “ectopic meningiomas” or “primary extradural meningiomas.”87,88 These are rare lesions that account for less than 2% of all meningiomas.88 Intraosseous meningiomas constitute 14% of primary extradural meningiomas.89
Several differences between primary extradural meningiomas and their more common intradural counterparts have been observed. The female preponderance of diploic meningiomas may not be as pronounced as it is with other meningiomas, although this finding is somewhat inconsistent.87,88 Second, although both intradural and ectopic meningiomas affect adults, only ectopic meningiomas have a second incidence peak in the pediatric population.88,90 Finally, extradural meningiomas may have a greater likelihood of being atypical or frankly malignant than intradural meningiomas do (11% rather than 2%).88,91
Intradiploic meningiomas are thought to arise from rests of arachnoid cap cells trapped within sutures at birth and during molding of the head.92,93 An alternative hypothesis is that ectopic meningiomas arise from abnormal differentiation of multipotent mesenchymal cells.87,88 Their cause may in fact be multifactorial.88 Awareness of a painless, slowly growing, palpable mass is the most common manifestation.87,88,90,94 Headaches and local tenderness are also possible.87,93 Lesions in the temporal bone may cause hearing loss or facial weakness.95,96 A case of multiple intraosseous meningiomas has been reported.97
Histologically, intraosseous meningiomas are not different from their intradural counterparts; they have a wide range of histopathologic findings and positive immunostaining for epithelial membrane antigen and vimentin.88,89,94
On plain radiography and CT, most intraosseous meningiomas are hyperostotic, although some are lytic or both lytic and sclerotic.87,89,90,92,95 There is some speculation that as the lesion progresses, it becomes more lytic and less sclerotic.92 CT is often the most helpful imaging modality because it allows assessment of the extent of bone involvement.87,89,94,96,98 CT and plain radiographs demonstrate expansion of the diploë, with the inner and outer tables of the skull separated and thinned over the biconvex mass.89,91,93 As seen with MRI, the tumors appear hypointense on T1-weighted and hyperintense on T2-weighted images.91 As with intradural meningiomas, intraosseous meningiomas display homogeneous contrast enhancement on both CT and MRI.89,91,98 On basal views of the skull, the foramen spinosum may be enlarged because of hypertrophy of the middle meningeal artery.92 Using angiography, the intradural arteries and dural sinuses are seen to be pushed away from the inner table of the skull; this is the opposite of what is found with intradural meningiomas, wherein the vessels are displaced toward the inner table.92 There may be a tumor blush from its branch feeders from the ECA.98
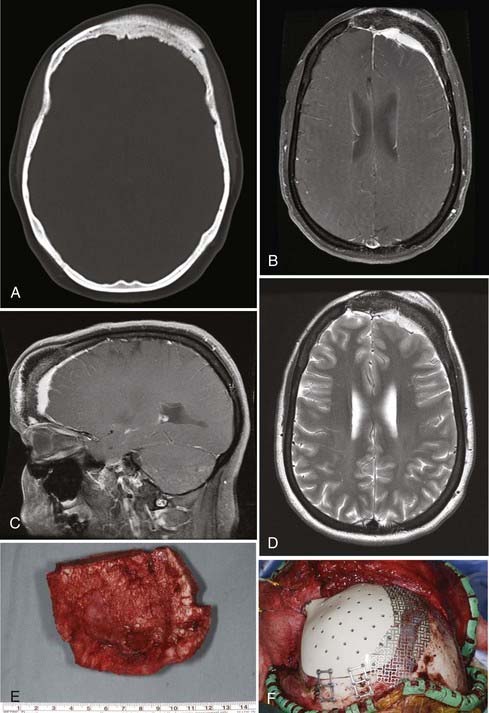
Intraosseous meningiomas of the skull are seldom correctly diagnosed preoperatively.87,88 Surgical resection is the treatment of choice for these lesions (Fig. 148-3). En bloc resection with margins, when feasible, may decrease the risk for recurrence.87,88,90,91,93,94,96
Malignant
Osteogenic Sarcoma
Osteogenic sarcoma (osteosarcoma) is the most common malignant tumor of bone, although it is relatively rare in the skull.99–103 In the skull, the cranial vault is involved more frequently than the base.102 These tumors may arise de novo or, more commonly, as a sequela of radiation treatment or another bone disorder such as Paget’s disease or fibrous dysplasia.1,10,100,101,103–106 Osteogenic sarcomas that develop secondary to radiation treatment are thought to be more aggressive and have a worse prognosis than those arising de novo.100 Additionally, de novo osteosarcomas tend to have a later onset.100
Osteosarcomas are generally manifested as localized, painless, expansile masses.1,100,102 Patients with tumors attaining large size or involving the skull base may complain of local tenderness, headaches, proptosis, ophthalmoplegia, facial weakness, decreased hearing ability, or tinnitus.100,101,103,106 Although extensive bony destruction can occur, the dura, brain, and venous sinuses are only rarely involved.10 The alkaline phosphatase level may be a useful diagnostic test; in a patient with known fibrous dysplasia, elevation of alkaline phosphatase may signify malignant transformation.100,103,105
Histologically, osteogenic sarcomas consist of a sarcomatous spindle cell stroma with an associated osseous component.1,101,103 The stromal cells demonstrate varying amounts of anaplasia and pleomorphism.1 Formation of bone or osteoid by the tumor cells is required for diagnosis.11,26 Necrosis may be present as well.99,102 A low-grade, well-differentiated subtype of osteosarcoma has been reported.101
Osteosarcomas may be either osteoblastic, osteolytic, or mixed, and their radiologic appearance is contingent on the relative amounts of each subtype.11,100,103 Osteoblastic lesions appear more sclerotic, and osteolytic lesions appear more radiolucent.103 Mixed, predominantly osteoblastic lesions are most common.100 CT is helpful in determining the amount of bony involvement.11,100 The classic appearance on radiography and CT consists of bony destruction, cortical expansion, and a “sunburst” periosteal reaction.101,104 CT may also demonstrate areas of irregular calcification, as well as low-attenuation areas representing necrosis.102 MRI generally demonstrates a heterogeneous signal on both T1- and T2-weighted sequences and may be helpful for determining the presence of intracranial invasion.11,100,102 Contrast enhancement is typically heterogeneous.101 A radionuclide bone scan may show increased uptake of radioactive material by the tumor.11,99 Angiography, when performed, demonstrates pathologic feeding of the lesion by branches of the ECA.100,102,106
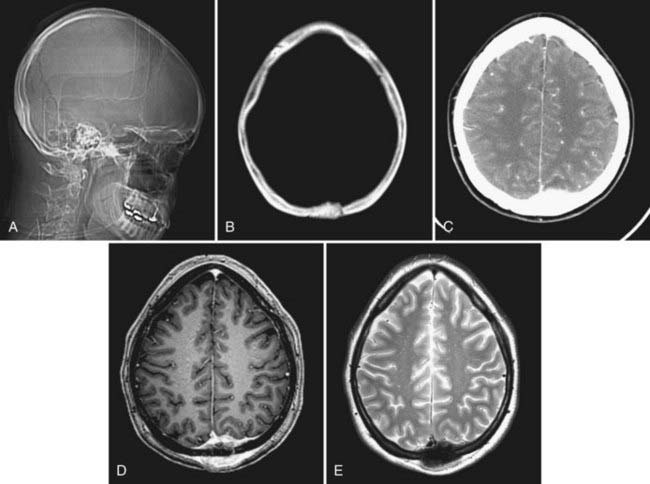
Gross total resection of osteosarcoma is the ideal treatment when safely attainable.99,102,103 Local tumor recurrence is common after partial resection (Fig. 148-4).99 Preoperative embolization may be a helpful surgical adjunct.102,106 Osteosarcomas are relatively radioresistant, although some surgeons recommend irradiation of any residual tumor after partial resection.99,100,105 Several groups have reported a dramatic increase in patient survival rate with the addition of chemotherapy.99,100,102,107 Salvati and coworkers reported an increase in the 2-year survival rate from 15% to 17% to 55% to 60% with the addition of chemotherapy.100 Chennupati and associates reported that a chemotherapeutic regimen of methotrexate, doxorubicin, and cisplatin increased the 5-year survival rate from 20% to between 60% and 70%.99 Sundaresan and colleagues advocated neoadjuvant treatment with this same regimen.107 The low-grade well-differentiated subtype of osteosarcoma is relatively chemoresistant.101
Although osteosarcomas of the skull tend to metastasize later than those arising in other primary sites, distant metastases will develop in 7% to 17% of patients.99 This figure is closer to 10% with the low-grade well-differentiated subtype, and the prognosis for this subtype is better.101 Although systemic metastases are a common cause of death in patients with osteosarcoma arising from the long bones, local recurrence is more commonly the cause of death in those who have skull osteosarcomas.106 Metastasis to extracranial sites should be regarded as a treatable rather than a terminal event.100 The lungs are the most common site of distant dissemination.99,100,102,106 Accordingly, post-therapeutic imaging surveillance should include chest radiography or CT.99 Spinal metastases have also been reported.106 Because some cases of osteosarcoma in the skull are, indeed, metastatic to that location from an extracranial primary site, a comprehensive survey of the body is mandatory with this disease (Fig. 148-5).
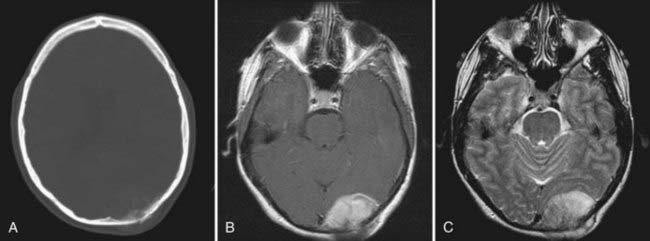
The extent of intracranial involvement by osteosarcoma at the time of diagnosis is thought to be the most important prognostic factor.102,106
Fibrosarcoma
Fibrosarcoma is a rare, malignant tumor that may arise from degeneration of a preexisting lesion such as a fibroma1 or from Paget’s disease,10,108 or it may occur after radiation treatment.10,108–110
Fibrosarcomas are typically indolent, asymptomatic masses,1,111 although lesions at the base of the skull may be associated with cranial nerve palsies.10 Extension into the dura or brain parenchyma is uncommon.10 Fibrosarcomas are the most common type of sarcoma to arise as a late complication of irradiation of pituitary adenomas.109,110
Histologically, these lesions are characterized by interlaced bundles of spindle cells and collagen fibers in a “herringbone” pattern.26,108 There is no associated osteoid or bone formation.108,109 Fibrosarcomas are very cellular malignant lesions with elevated mitotic activity.26,108,109 They can generally be divided into three categories: low grade (most differentiated), moderately differentiated, and poorly differentiated.1 Cellularity and mitoses are both increased in the latter category.1,108,109 Necrosis may be present as well.10,109 Degeneration of a low-grade fibrosarcoma to a more malignant osteogenic sarcoma has been reported.109
Fibrosarcoma is often seen on imaging studies as a lytic lesion with cortical destruction or expansion, or both, and soft tissue extension.11,108,111 These tumors are generally periosteal in origin.10 Fibrosarcomas are frequently radiolucent because there is no bone formation or calcification.108 Alkaline phosphatase levels are not typically elevated, which may help distinguish this lesion from osteogenic sarcoma preoperatively.108 However, CT may demonstrate extensive bony destruction.111 MRI is helpful in determining the presence of intracranial extension.111 Peripheral contrast enhancement of the tumor may be noted on CT or MRI.111 Angiography may demonstrate tumor feeding by branches of the ECA.111
Fibrosarcoma appearing in childhood, including the congenital subtype, is a somewhat distinct clinical and histologic entity.111 Childhood fibrosarcoma tumor cells are more primitive and rounded and less pleomorphic than the adult type.111
En bloc surgical resection is generally the treatment of choice in both adults and children when feasible.108,111,112 Neoadjuvant chemotherapy or irradiation, or both, may be necessary when en bloc resection is not attainable.111,112 In adults, good tumor control has been reported with radiation therapy after incomplete tumor resection.113
Chondrosarcomas and Chordomas
Chondrosarcomas and chordomas are rare, slow-growing, infiltrative tumors of the skull base.114–117 They are pathologically distinct entities, although both have a natural history of indolent expansion, local invasion, and relentless recurrence.114,116,118 Chordomas are more common and tend to have a worse prognosis than low-grade chondrosarcomas.114,116,118,119 The 5-year survival rate in one series was reported to be 90% for chondrosarcomas and 65% for chordomas of the skull base.115 Chordomas are more commonly found at the midline in the clivus and cause brainstem compression, whereas chondrosarcomas are typically found away from the midline and give rise to cranial neuropathies.118,120,121 The chordoma subtype “chondroid chordoma” may be difficult to distinguish pathologically from low-grade chondrosarcoma, and such distinction is a matter of controversy.10,118
Chondrosarcoma
Chondrosarcoma is a malignant neoplasm of cartilage.1,10,116 The histologic origin of this tumor is unclear, and it originates either from primitive mesenchymal cells or from embryonic rests of the cartilaginous matrix near fused joints such as the petroclival fissure.114,116,118,120
Chondrosarcomas may cause extensive bone destruction, in addition to invading the middle and posterior fossae.10 Patients typically have headaches and cranial neuropathies, particularly abducens palsy.10,114,116,120 Lesions of the skull convexity may be manifested as painless expanding masses.1 Although chondrosarcomas typically arise de novo, progression from chondroma to chondrosarcoma has been documented.10,26,58 Additionally, multiple enchondromatosis type 1 (Ollier’s disease) predisposes patients to the development of chondrosarcoma.122
The malignant potential of chondrosarcoma is variable,10 but regardless, the prognosis is generally poor as a consequence of its relentless tendency for local recurrence.1
Chondrosarcomas are subclassified as “myxoid” (“conventional low grade”), “dedifferentiated,” and “mesenchymal,” with the myxoid type being the most common.114,118 Histologically, chondrosarcomas are hypercellular with hyperchromatic and pleomorphic nuclei.1 Multinucleate cells may be present.1 The hypercellular small cell component is interspersed with islands of hyaline cartilage.26 Histologically, the small cell element closely resembles hemangiopericytoma, with staghorn vasculature and intercellular reticulin staining.26
At times it may be difficult to differentiate chondrosarcoma from chondroid chordoma, particularly when the tumor arises from the midline.26,115 Unlike chondroid chordoma, however, chondrosarcomas are nonreactive for keratin and epithelial membrane antigen.26,114
On MRI, chondrosarcomas are usually lobulated lesions that appear isointense to hypointense on T1-weighted images and hyperintense on T2-weighted images and show heterogeneous contrast enhancement.116,119 This appearance is very similar to that of chordomas, but chordomas are more commonly found in the area of the clivus, whereas chondrosarcomas are more typically located off the midline.116,119 CT and radiography may demonstrate calcifications and ossifications within the tumor mass.11 Angiography may reveal extensive vascularity and an attendant tumor blush in higher grade lesions.11
Surgical resection followed by adjuvant radiotherapy is the treatment of choice for chondrosarcomas of the skull.114,115,120 The goal of gross total resection is often difficult to achieve because these tumors infiltrate the critical structures of the skull base.117 Even with multimodality therapy, recurrence is the norm and translates into a poor overall survival interval.119
Although chondrosarcomas are generally believed to be relatively radioresistant, some form of radiation treatment is typically part of the therapeutic regimen.116,119
Two groups have recently reported good results with the use of stereotactic radiosurgery (SRS) as a postoperative adjunct for these lesions.117,120 Using postoperative SRS for residual or recurrent chondrosarcoma, Martin and coauthors reported a 5-year local control rate of 80% (±10%).120 They further speculated that SRS may be used in some cases as the primary treatment modality without surgery.120 Hasegawa and colleagues found that SRS was a useful postoperative adjunct, provided that the residual tumor volume was less than 20 mL.117
al-Mefty and coworkers reported good results without the use of postoperative radiotherapy in cases in which gross total resection was achieved and the tumor was found to be of the conventional low-grade histologic type.118 They did caution that this would not be the ideal treatment for patients with the dedifferentiated or mesenchymal types.119
Proton beam adjuvant therapy may be helpful, but ideal dosing for chondrosarcoma is still being determined.114,119
Chordoma
Chordomas are slow-growing tumors that arise from notochordal remnants.34,123–125 Chordomas of the skull most commonly occur at the skull base, with most arising from the clivus.1,121 These tumors may occur at any age, with the mean age being 36.9 years.1,11,116 As with skull base chondrosarcomas, most patients with chordomas have an abducens palsy and attendant diplopia. Headaches and lower cranial nerve palsies are also common findings.1,116,123,126 Pituitary dysfunction, torticollis, and cerebellar signs have been reported.121 Males are affected more frequently than females by a ratio of 2 : 1.11,127 These lesions typically form bulky, soft gray masses compressing the base of the brain and cerebellum.10 Although histologically benign, these tumors are locally invasive and generally portend a poor long-term prognosis.10,124 Untreated, a patient with a clival chordoma rarely survives for more than 30 months.128 Five- and 10-year survival rates with present treatment regimens are estimated to be 50% to 80% and 35%, respectively.127,129 However, the biologic behavior of these lesions is quite variable.129 Being female, experiencing tumor necrosis before radiation therapy, and having a tumor volume greater than 70 mL are all independent predictors of shortened overall patient survival.130 Some chordomas may be frankly malignant with metastatic potential.10,124 Metastasis is relatively uncommon, however, and occurs in 10% to 18% of patients and generally late in the clinical course.124,125 The danger arising from these “benign” tumors lies in their relentlessly recurrent, locally aggressive nature in the region of the vital structures of the skull base.123,125
Common chromosomal abnormalities include gains at specific regions of chromosomes 1, 7, and 19, as well as polysomy of chromosome 7.127,131 Immunohistochemical studies show that chordomas stain for S-100 protein, cytokeratin, and epithelial membrane antigen—the latter of which may be particularly helpful in distinguishing chordoma from chondrosarcoma.10,116,126
In gross appearance, chordomas are lobulated, gelatinous, semitranslucent gray masses.124,126 Histologically, chordomas are characterized by physaliphorous (“bubble-bearing”) cells, which also distinguishes them from chondrosarcomas.34,116 Cellular pleomorphism, mitoses, and hyperchromatic nuclei are uncommon and have little correlation with patient survival.124,125 Two percent to 8% of chordomas may contain regions of malignant mesenchymal tissue and are appropriately designated “dedifferentiated chordomas.”10,132
The classic finding on radiographs of skull chordomas is an expansile, lytic lesion of the clivus with periosteal elevation.124 Both soft tissue and osseous components are typically present.124 Chordomas usually appear isointense to hyperdense with variable heterogeneous contrast enhancement on CT.11 On MRI, chordomas appear isointense to hypointense on T1-weighted and hyperintense on T2-weighted images, with moderate to avid contrast enhancement.11,116 CT is useful for evaluation of bony destruction and tumor calcification—the latter being somewhat uncommon relative to chordomas of the sacrum.11,124,125 Angiography may be helpful in delineating the relationship of the tumor to the petrous carotid artery or the vertebral artery.126 Encasement or displacement of vascular structures may be noted, but significant stenosis is infrequent.11,129,133 The vascularity of the chordomas themselves is quite variable and may or may not demonstrate a blush on angiography.11,126 Again, the overall appearance is similar to that of chondrosarcomas, although chondrosarcomas are more typically found away from the midline.116
Surgical excision plays an important (and generally primary) role in the management of clival chordomas.123,126,129,132,134,135 Extensive resection has been associated with prolonged survival, but this must be balanced against the risk for increased morbidity with surgery.123 True “oncologic” resection is rarely achieved because of the invasive character of chordomas and their proximity to critical vessels and nerves.136 Although often described as soft and gelatinous, chordomas may be tough and fibrous at surgery; both consistencies can present a surgical challenge.137
Surgical approaches to clival chordomas include fronto-orbitozygomatic, zygomatic–extended middle fossa, transmaxillary, transcondylar, transbasal,123,138 and the transnasal, transsphenoidal approach, with or without endoscopic visualization.123,126,139 Additionally, DeMonte and associates described a transmandibular circumglossal retropharyngeal approach.137 Harsh and colleagues described a pedicled rhinotomy for a midface transnasal approach.121 Based on their location, al-Mefty and Borba divided chordomas into those that required only one skull base surgical approach (most cases in their series) and those that required combined approaches.123 Harsh and colleagues divided chordomas into those in the upper third of the clivus, which are best reached by a transnasal transsphenoidal approach; those in the lower third of the clivus, which require a transoral approach; and those lateral to the internal carotid artery or in the medial petrous bone, which require an additional lateral approach.121
At surgery, the tumor may be entirely extradural or may have eroded through the dura to become both extradural and intradural.121,126 The amount of intradural tumor may influence the surgical approach used.133 al-Mefty and Borba stressed the importance of drilling out the involved bone, as well as resecting the soft tissue mass.123 “Seeding” of the operative site with chordoma cells at surgery can occur unless the utmost care is taken during removal.140 Additionally, there is one reported case of an abdominal fat graft harvest site becoming seeded with chordoma.140 Leakage of CSF and cranial neuropathy are among the most common surgical complications.128,133,135 In experienced hands, most cranial nerve deficits associated with surgery tend to resolve with time.128
Because of the poor results obtained by treating chordomas with conventional radiotherapy, they were initially thought to be “radioresistant” tumors. The advent of more conformal, high-dose irradiation, particularly proton beam irradiation, has led to the standard use of postoperative radiotherapy.116,121,123,124,135,136 Surgical resection has been performed after irradiation but is typically more difficult because normal tissue planes are disrupted, which places vessels and nerves at greater risk.126,133,134 There is some disagreement regarding whether radiation therapy should be used in all cases postoperatively, when there is known residual tumor, or only at recurrence.121,123,128 A dose of at least 60 Gy is required for therapeutic effect.129 As it has become more available, proton beam radiotherapy is more commonly being used for residual or recurrent tumor.119,123,128,135 One large retrospective series found a 5-year survival rate of 79% for chordoma patients treated with proton beam radiotherapy after surgical resection.119 As with the Bragg peak of proton beam radiotherapy, SRS provides the ability to direct a high concentration of radiation to a well-demarcated area.129,135,138 Good results have been reported with the use of SRS for recurrent or residual disease.120,141 Additionally, Martin and colleagues have used radiosurgery as primary treatment in two patients.120
Chemotherapy is generally ineffective for the treatment of chordomas but may have a role in treatment of the dedifferentiated chordoma subtype.132
Recurrences of chordoma are common, but survival for as long as 46 years has been demonstrated in a poorly defined subset of patients.129,136 The risks associated with surgery are greater in patients who have previously undergone surgery or radiation therapy, or both.133,134

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


