Spasticity and Movement Disorders
Although thousands of children have spasticity or other movement disorders, few pediatric neurosurgeons are involved in their care. That under-involvement partially reflects the paucity of neurosurgical treatments that were available historically. Until 1987, when Warwick Peacock and colleagues popularized selective dorsal rhizotomies,1 pediatric neurosurgeons had little to offer children with spasticity. Since 1987, several methods for treating spasticity have become available, including oral medications (tizanidine), intramuscular (IM) medications (botulinum toxins), intrathecal medications (baclofen), and nonselective rhizotomies. Surgical options for treating dystonia and other movement disorders have increased with the introduction of intrathecal baclofen and deep brain stimulation. These options allow pediatric neurosurgeons to become involved in the care of a large group of children with whom we previously had minimal involvement, children whose quality of life can often be improved substantially.
The under-involvement of pediatric neurosurgeons in the care of children with spasticity or other movement disorders may also reflect a preference for not being involved in the treatment of severely disabled children and an uncertainty about how best to treat these complex patients. Much of their complexity is due to the fact that they often have more than one movement disorder; combinations of spasticity and dystonia, or of chorea and athetosis, are common, particularly in children with cerebral palsy (CP), and the most frequent cause of movement disorders in children is CP. Video examples of pediatric movement disorders are available on the WeMove Web site, http://www.wemove.org.
76.1 Spasticity
A task force defined spasticity as hypertonia in which one or both of the following signs are present: (1) resistance to externally imposed movement that increases with increasing speed of stretch and varies with the direction of joint movement, and/or (2) resistance to externally imposed movement that rises rapidly above a threshold speed or joint angle.2 Clinically, spasticity may be defined as a velocity-dependent, increased resistance to passive muscle stretch. Strictly speaking, spasticity does not cause sustained, tonic increases of muscle tone, but rather increases in tone that vary according to movement, alertness, pain, and anxiety of the patient. Spasticity may be considered an isokinetic movement disorder because the amount of movement is not increased, as it is in hyperkinetic disorders like dystonia and chorea.
Spasticity is classified according to the limbs affected: spastic quadriparesis if all extremities are affected; spastic diplegia (or spastic paraparesis—the terms are synonymous) if the lower extremities are involved; spastic hemiparesis if an ipsilateral arm and leg are involved; and spastic monoparesis if only one limb is affected. Spasticity can affect trunk and cervical muscles, but no classification system grades their involvement. In Europe, children with spasticity that affects all extremities equally are said to have spastic tetraplegia, whereas those with more severe involvement of the lower extremities than the upper are said to have spastic diplegia.
Spasticity has enormous clinical and economic implications. If untreated, it leads to progressive disability, discomfort, and deformity, and to multiple orthopedic operations to correct the deformities.
76.1.1 Epidemiology
Spasticity is common. It occurs in approximately 60% of individuals with CP (which occurs in 2.5 of 1,000 live births) and thus affects some 300,000 individuals younger than than 18 years in the United States. The incidence of CP is not declining in the United States, probably because of the increased survival of low-birth-weight infants; the risk for CP is increased 25 to 30 times for those weighing less than 1,500 g.3 Spasticity also occurs in substantial numbers of children after serious head injuries and strokes, but the number with spasticity from those causes is unknown.
When spasticity occurs in association with CP, it is often not identified until 1 year of age or more. It may be more severe in the first 2 to 3 years of life, then improve as myelination increases. In a landmark study, Nelson and Ellenberg4 found that approximately two-thirds of children in whom spastic diplegia was diagnosed—and half of all children in whom CP was diagnosed at their first birthday—“outgrew” or lost the motor signs of the CP by their seventh year. Spasticity in CP may improve over time, and it does not worsen, although it may appear to do so for three reasons: increasing muscle strength, worsening contractures, and misdiagnosis (when worsening hypertonicity is due to dystonia rather than to spasticity). Spasticity that occurs in the first months after severe head injury is particularly likely to improve, so that permanent treatments, such as neurectomies, are infrequently appropriate during that year (although reversible treatments, such as botulinum toxin injections and intrathecal baclofen, may be appropriate).
76.1.2 Pathology and Pathophysiology
The pathophysiology of pediatric movement disorders was reviewed by Sanger in 2003.5 Muscle tone (the state of muscle contraction) is regulated by output from α motor neurons and is influenced by competing impulses: excitatory impulses, which enter the spinal cord via type Ia afferents from muscle spindles and cause the release of excitatory neurotransmitters, such as glutamate and aspartate; and descending inhibitory impulses from the basal ganglia and cerebellum, which cause the release of the inhibitory neurotransmitter γ-aminobutyric acid (GABA).6 Spasticity in humans is not due to increased gain of the muscle spindle, increased excitation of type Ia afferents, or increased γ efferents. There is probably reduced reciprocal inhibition of antagonistic motor pools by Ia afferents and decreased nonreciprocal inhibition by Ib terminals.7 Spasticity that develops in childhood differs somewhat from spasticity that develops in adults; early central nervous system (CNS) injury seems more likely to result in a reorganization of corticospinal projections that leads to co-activation of agonist and antagonist muscle groups.8,9
In simplistic terms, virtually all spasticity—whether of cerebral or spinal origin—can be attributed to an imbalance of excitatory and inhibitory impulses, with a relative deficiency of GABA in the spinal cord because of injury either to the developing brain or to the spinal cord conducting the descending inhibitory impulses. This pathophysiologic model also suggests two potential treatments: reducing the afferent excitatory input by rhizotomy and increasing the inhibitory neurotransmitter with a GABA agonist, such as baclofen. The paradigm also gives rise to the postulate that descending inhibitory impulses might be increased by cerebellar or spinal cord stimulation with implanted electrodes, treatments that were not effective.10,11
76.1.3 Clinical Features and Grading
Symptoms of spasticity include muscle stiffness and tightness, fatigability, and pain. Pain often occurs during the night as a result of lower extremity muscle cramps or the inability to change positions. Spasticity in proximal lower extremity muscles causes progressive hip dislocation, which leads to acetabular deformity; this in turn may later cause intractable hip pain.
Spasticity typically involves certain muscles more than others; flexors and internal rotators are affected more than their antagonists. For children with spastic quadriparesis, that pattern of involvement results in the common stance of flexion at the elbows and wrists, with the child standing on the toes, the knees and hips somewhat flexed, and the legs internally rotated.
Spasticity associated with spinal cord injuries differs from cerebral spasticity in two ways: it is typically more severe because more descending inhibitory impulses are abolished, and it is more likely to be associated with painful muscle spasms. After spinal cord injuries, Ashworth Scale scores of 4 to 5, severe clonus, and muscle spasms are typical, whereas scores of 3 to 4 and less severe clonus are seen in cerebral spasticity. Clonus may be present at the ankle or knee and may occur spontaneously.
On examination, spasticity is usually evident exactly as it is defined, as a velocity-dependent, increased resistance to passive muscle stretch, so that as a joint is passively moved at varying rates, resistance varies. Spasticity is graded most commonly with the Ashworth Scale or Modified Ashworth Scale (▶ Table 76.1), based on the subjective grading of resistance to passive muscle stretch. Interrater reliability of the scale has been demonstrated.15 A less common method of evaluating tone is with the Tardieu Scale, although it is thought to provide a more accurate assessment than the Ashworth Scales (▶ Table 76.2).16 Spasticity is usually assessed with the child lying down, although a few children have dynamic spasticity, in which their tone is normal when they are lying down but increases when they stand or walk, especially tone in the adductors and plantar flexors.
| Ashworth Scale | Modified Ashworth Scale | ||
| Score | Degree of muscle tone | Grade | Description |
| 1 | No increase in muscle tone | 0 | No increase in muscle tone |
| 2 | Slight increase in muscle tone, giving a “catch” when affected part is moved in flexion or extension | 1 | Slight increase in muscle tone |
| 3 | Moderate increase in muscle tone, passive movement difficult | 1+ | Slight increase in muscle tone, manifested by a catch, followed by minimal resistance throughout the remainder (less than half) of the range of motion |
| 4 | Considerable increase in muscle tone, passive movements difficult | 2 | More marked increase in muscle tone through most of the range of motion, but affected part(s) easily moved |
| 5 | Affected part rigid in flexion or extension | 3 | Considerable increase in muscle tone, passive movement difficult |
| 4 | Affected part(s) rigid in flexion or extension | ||
| Velocities | |
| V1 | As slow as possible, slower than the natural drop of the limb segment under gravity |
| V2 | Speed of limb segment falling under gravity |
| V3 | As fast as possible, faster than the rate of the natural drop of the limb segment under gravity |
| Scoring | |
| 0 | No resistance throughout the course of the passive movement |
| 1 | Slight resistance throughout the course of the passive movement, no clear catch at a precise angle |
| 2 | Clear catch at a precise angle, interrupting the passive movement, followed by release |
| 3 | Fatigable clonus with less than 10 seconds when the pressure is maintained and appearing at the precise angle |
| 4 | Unfatigable clonus with more than 10 seconds when the pressure is maintained and appearing at a precise angle |
| 5 | Joint immobile |
| Note: This test is performed with the child in the supine position, with the head in midline. Measurements are taken at three velocities: V1, V2, and V3. Responses are recorded at each velocity as X/Y, with X indicating the 0 to 5 rating and Y indicating the the angle (in degrees) at which the muscle reaction occurs. If the limb is moved at different velocities, the response to stretch can be more easily gauged because the stretch reflex responds differently to changes in velocity. | |
When a child with spasticity is evaluated, it is important also to test the strength of the affected muscles because some children supplement weak voluntary muscle contractions with the involuntary muscle contractions of spasticity, so that gait and pivot transfers may worsen if spasticity is reduced. In children with spastic lower extremities, leg strength can be assessed by having them rise from a sitting position on a stool, by having them squat and rise in increments, and by having them stand on one foot at a time (Trendelenburg test); pelvic tilt to the lifted side indicates hip abductor weakness of the leg they are standing on.
If a muscle is affected by spasticity for several months, the muscle and its tendon undergo a permanent shortening—a musculoskeletal contracture—which is a fixed deformity that does not improve after treatment of the spasticity. Ideally, spasticity should be treated early to prevent the development of contractures, which occur most commonly in muscles that are affected most by spasticity: foot plantar flexors, knee flexors, and hip adductors.
Pediatric neurosurgeons who treat children with CP occasionally encounter some with hereditary spastic paraparesis (Strümpell-Lorrain disease), which is often misdiagnosed as CP.17 Hereditary spastic paraparesis is characterized by no history of perinatal asphyxia, normal intellect, slightly delayed motor milestones, and slowly progressive spastic paraparesis. A careful family history often uncovers symptoms in family members, although the extent of involvement may vary considerably from member to member. Neurosurgeons are also asked to see children in whom spastic CP has been misdiagnosed, but who have dystonia.
76.1.4 Diagnostic Studies
No computed tomographic (CT) or magnetic resonance (MR) imaging characteristics are pathognomonic for spasticity, although MR images of children with spastic diplegia frequently demonstrate periventricular leukomalacia (▶ Fig. 76.1), and those of children with spastic quadriplegia often demonstrate multifocal, cystic encephalomalacia that involves both the cortex and underlying white matter. Neuroimaging features of spasticity and other movement disorders have been described.18
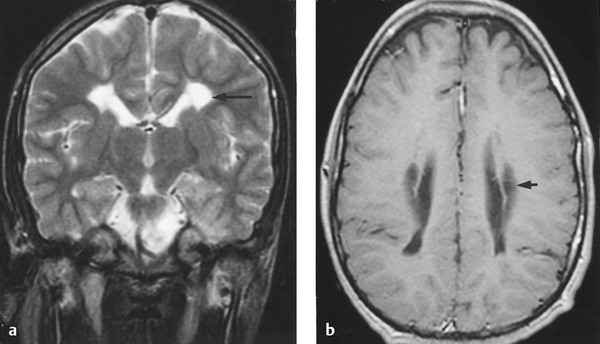
Fig. 76.1 (a) Axial and (b) sagittal magnetic resonance images demonstrating periventricular leukomalacia in a pediatric patient with spastic diplegia.
No methods are widely available to quantitate spasticity. Thus far, neurophysiologic techniques, such as H-reflex excitability curves and devices to measure velocity, joint angle, and torque, have not gained widespread clinical use. Electromyograms do not quantitate spasticity. In gait analysis studies, electrodes are applied to multiple lower extremity muscles; joint markers are applied to the hips, knees, and ankles; and children are videotaped while walking. Gait analysis allows a determination of which muscles contract during the gait cycle (frequently identifying the simultaneous contraction of agonists and antagonists), stride length, cadence, and joint angles during the gait cycle. The interpretation of gait analysis is complex, but the resultant information is often helpful in planning subsequent orthopedic and neurosurgical intervention in ambulatory children. A complete discussion of gait analysis is beyond the scope of this chapter but is available in a publication.19
76.1.5 Treatment
It is important to remember that not all spasticity needs to be treated; spasticity that is mild, functionally useful, and neither interfering with function nor causing contractures can be left alone. If spasticity is to be treated, the treater and the treated should agree on the treatment goals. Common goals are to (1) improve function, (2) facilitate care, (3) retard or prevent the development of contractures, and (4) reduce or prevent pain. Patients commonly, and not unexpectedly, have higher hopes and expectations of treatment than do the treating physicians, despite careful preoperative discussions.
Spasticity can be treated with oral medications, intramuscular injections, intrathecal medications, and neurosurgical operations. The plethora of treatment options increases the dilemma for parents and patients and, to some degree, for physicians. Few centers have experience with all treatment modalities. Almost no good-quality studies have been done to evaluate the results of individual treatment modalities, much less to compare one modality with another. Ideally, children with spasticity should be evaluated in a multidisciplinary clinic that has experience in all treatment modalities. A gross motor function classification system (GMFCS) has been developed to classify function in children with CP so that the effects of treatments can be more readily compared.19
Treatment selection also varies according to the severity of the spasticity, the extent of the muscles involved, the age of the child, and the therapeutic goals, as discussed later. ▶ Table 76.3 lists the common treatments and their general indications.
| Method | Age (y) | Diagnosis | Characteristics | Expected results | Follow-up care | Outcome | Side effects, risk |
| Oral medications | Any age, most often 2 to 5 | Spastic quadriplegia, traumatic brain injury | Diffuse spasticity | Mild decrease in spasticity in arms and legs | PT, OT as needed | SPRs or ITB often needed later | Drowsiness |
| Botox injections | Any age, less often older than 10 | Spastic diplegia, spastic quadriplegia | Isolated spasticity; too young for ITB, SPRs | Decrease in spasticity in injected muscle(s) for 2 to 4 months | PT, OT to increase range of motion and to increase strength | Improved gait, sometimes improved arm function | None with usual doses |
| Rhizotomy (SPRs) | 4 to 8 (most common), rare after 16 | Spastic diplegia or quadriplegia, capable of ADLs | Good leg strength, no severe contractures, motivation for PT | Marked, permanent, nonadjustable decrease in spasticity | Extensive PT, OT | Improved walking, improved ADLs, decrease in orthopedic operations | Infection, 2%; wound, 1%; CSF leak, 3% |
| Older than age 3, before multiple contractions | Spastic diplegia or quadriplegia, not capable of ADLs | Severe leg spasticity interfering with care | Minimal | Easier care | Infection, 2%; wound, 1%; CSF leak, 3% | ||
| Baclofen (ITB) | Older than age 3, big enough to insert pump | Spastic quadriplegia, spasticity in legs greater than or equal to spasticity in arms, capable of ADLs | Severe spasticity, positive response to test dose, spasticity limiting function | Adjustable decrease in spasticity | Frequency of PT, OT depends on goals | Improved walking, improved ADLs, improved speech, decrease in orthopedic operations, easier care | Infection, 5 to 10%; wound, 5 to 10%; CSF leak, 5 to 10% |
| Spastic quadriplegia, not capable of ADLs | Spasticity interfering with care | Minimal PT | Easier care | ||||
| Posttraumatic brain injury | Severe spasticity in arms or legs, usually more than 1 year after injury | PT, OT for range of motion | |||||
| Abbreviations: ADLs, activities of daily living; CSF, cerebral spinal fluid; ITB, intrathecal baclofen; OT, occupational therapy; PT, physical therapy; SPRs, selective posterior rhizotomies. | |||||||
Oral Medications
Three oral medications are thought to be useful in treating pediatric spasticity: baclofen, diazepam, and dantrolene12,20; all act systemically and decrease spasticity diffusely. No study has compared their effectiveness, which is usually mild and often seems to wane over a few years. Medication dosages are listed in ▶ Table 76.3. For each medication, doses are adjusted to obtain the greatest therapeutic benefit with the fewest side effects. The use of oral medications for treating spasticity has been reviewed recently.22
Baclofen (Lioresal; Novartis Pharmaceuticals, East Hanover, NJ), or chlorophenol GABA, is a GABA agonist that was synthesized to be an anticonvulsant (its antiepileptic activity was minimal). It is absorbed well and rapidly from the gastrointestinal tract, with peak serum levels 2 hours after a dose. The serum half-life is 3 to 4 hours. Baclofen is a racemic mixture whose site of action is the spinal cord, where primarily the levo-enantiomer acts both to decrease the release of excitatory neurotransmitters from type Ia fibers in superficial (Rexed layers I and II) layers of the spinal cord and to reduce postsynaptic impulses. Baclofen crosses the blood–brain barrier poorly; oral doses of 60 to 90 mg/d are usually associated with serum levels of 68 to 650 ng/mL, but with cerebrospinal fluid (CSF) levels of 12 to 95 ng/mL.23 In a double-blinded crossover study of 15 children with spasticity and GMFCS levels of IV or V, children receiving baclofen scored significantly better on the goal attainment scale than those receiving placebo.24
Diazepam (Valium; Roche Pharmaceuticals, Nutley, NJ) is a benzodiazepine receptor agonist that is also absorbed well and rapidly after oral administration. It crosses the blood–brain barrier moderately well and attaches to GABAA and GABAB receptors in the brain and spinal cord, so that it has a higher frequency of CNS side effects. Its half-life is 36 hours. It improves spasticity of children with CP, but effective doses frequently cause unacceptable sedation.25 Dependence may develop after chronic administration, so that the drug should be discontinued slowly, often over months.
Tizanidine (Zanaflex; Acorda Therapeutics, Ardsley, NY), an α2-agonist, appears to modulate the release of excitatory neurotransmitters from interneurons and afferent terminals. Tizanidine was approved for the treatment of spasticity based on the results of clinical trials of adult patients with spasticity from spinal cord injury or multiple sclerosis, in whom its effectiveness appeared to be similar to that of baclofen. A multicenter study of tizanidine in children with spasticity secondary to cerebral spasticity was recently concluded; the results have not been published as of September 2013.
Modafinil (Provigil; Cephalon, Frazer, PA) is a centrally acting stimulant that is thought to stimulate anterior hypothalamic nuclei. It was reported to improve spasticity in children with CP, but a subsequent double-blinded crossover study found that modafinil did not reduce spasticity or have a positive effect on quality of life.26
Dantrolene (Dantrium; Procter & Gamble Pharmaceuticals, Cincinnati, OH) is the only medication to act at a non-CNS site, the sarcoplasmic reticulum, where it prevents the calcium influx necessary for muscle contraction and thus is less frequently associated with drowsiness or other CNS side effects. Its peak activity is 4 to 8 hours after administration. Although dantrolene has been reported to cause hepatotoxicity, usually after 3 to 12 months of treatment, severe hepatotoxicity rarely occurs in children younger than 10 years old, and fatal hepatotoxicity has not been reported in patients younger than 20 years old.27
Baclofen is usually the first choice of oral agents. Doses are increased until improvement occurs, unacceptable side effects occur, or the maximum dose recommended for body weight is reached. If spasticity cannot be treated effectively with baclofen, it probably cannot be treated with other available agents used singly. Diazepam or dantrolene is occasionally given in combination with baclofen. Data comparing monotherapy with combination therapy are not available.
Neuromuscular Blockade
Neuromuscular blockade can be used to weaken muscle contractions, and thus spasticity, by interfering with nerve transmission, the neuromuscular junction, or muscle contraction. Medications available for IM injection include the following: botulinum toxin serotype A (Botox [Allergan, Irvine, CA]; Onabotulinumtoxin A—a purified version containing no extraneous proteins [Allergan]; Dysport [Ipsen, Slough, UK]); botulinum toxin serotype B (Myobloc; Solstice Neurosciences, South San Francisco, CA); phenol; and alcohol.28,29 No studies have compared their effectiveness. None treats spasticity per se; rather, they weaken the contraction of the injected muscle. IM injections are most appropriate for spasticity that affects localized muscle groups (e.g., plantar flexors and adductors) and are not appropriate treatment for generalized spasticity. However, for children with generalized spasticity, IM injections into the most severely affected muscles can be combined with oral medications that treat less involved muscles. Because the effects of IM medications usually wear off in a few months, such injections must be repeated and rarely provide a definitive treatment of spasticity. They are given with several goals: to decrease the spasticity of certain muscles (e.g., adductors) so that physical therapy can strengthen the antagonist muscles (abductors), to facilitate the serial casting of joints, and (rarely) to help determine if function of a limb would improve if spasticity were reduced. The use of injectable neuromuscular blockade to treat spasticity and other movement disorders was recently reviewed.30,31
Botulinum toxin acts at the neuromuscular junction to prevent the release of acetylcholine. There are seven botulinum neurotoxins (A through G), two of which are commercially available. Botox is type A, the most potent serotype; Myobloc is type B. Botox is injected directly into muscle; its effect commences within 1 to 3 days, is maximal at 21 days, and wanes—perhaps because of the development of new neuromuscular junctions—by 3 to 4 months afterward, when the injections can be repeated. Doses depend on the size of the muscle to be injected and on the severity of its spasticity. In the lower extremities, Botox is often injected into spastic gastrocnemius, hamstring, and adductor muscles. If injections are needed into muscles that are difficult to locate by palpation, such as the pronator teres or posterior tibial muscle, electrical stimulation will identify the desired injection site.
The maximal recommended Botox dose at present is 10 U/kg, although I, and others, have used 20 to 25 U/kg without adverse effects. The drug comes as a precipitate in vials of 100 U, is diluted to 10 U/0.1 mL or 10 U/0.2 mL, and injected into muscles at one to several sites. Dysport, the form of botulinum toxin available in the United Kingdom, is now available in the United States. It has approximately one-third the potency of Botox, and doses are accordingly higher.
The effects of Botox—alone or in combination with occupational therapy–on the upper extremities of children with spastic CP were evaluated in a 2010 Cochrane Systematic Review of 10 randomized controlled trials (RCTs).30 The combination of Botox and occupational therapy was more effective than occupational therapy alone in reducing impairment and improving activity-level outcomes, but not for improving quality of life. There was moderate evidence that the use of Botox alone was not effective. The effects of Botox on lower extremity spasticity and gait have been evaluated in recent reviews of RCTs. Boyd and Hays reviewed 10 RCTs and concluded that Botox had a moderate, dose-dependent effect on gait and lower limb function.31 Koog and Min reviewed 15 RCTs and concluded that botulinum toxin serotype A (BtxA) was little better than a sham control.32 Ryll et al reviewed 8 RCTs that compared BtxA plus usual care or physiotherapy versus physiotherapy alone.33 The studies showed moderate evidence for improved functional outcomes—mainly walking—at 2 to 24 weeks for those receiving BtxA, but no difference between BtxA and casting. BtxA treatment of ambulatory children with spastic CP has been shown to improve gait analysis parameters and to reduce oxygen consumption.34
Treatment with IM injections provides temporary improvement of spasticity and thus may be considered as a bridge to more definitive treatments. I often use Botox repeatedly in patients from age 2 years until age 5 or 6 years, by which time it is usually apparent whether the child will need longer-term treatment, such as a rhizotomy or an intrathecal baclofen pump. Only a few children who are treated with Botox in the 2- to 5-year interval improve enough that no subsequent intervention is needed. Children almost never develop immunity to Botox; the preparations that have been available in recent years are more purified and lack the protein to which early subjects occasionally developed antibodies.
Botox is tightly bound to the neuromuscular junction and a minimal amount enters the systemic circulation, so that systemic side effects are uncommon after injections into extremity muscles. Those effects include flulike symptoms, generalized weakness, dysphagia, and aspiration. In 2008, the Food and Drug Administration (FDA) reported that children treated for spastic CP had rarely developed serious side effects consistent with botulism.35 In a subsequent meta-analysis of 20 RCTs and 882 subjects who had received BtxA, a variety of symptoms were reported, including muscle weakness, urinary incontinence, fever, falls, and pain.36 Two deaths were reported but were thought to be unrelated to BtxA, and the authors concluded that BtxA had a good short-term safety profile. Despite the FDA warning, the use of BtxA remains widespread.
One circumstance in which Botox injection is probably underused is in children with severe head injury in whom gastrocnemius spasticity causes severe plantar flexion of the feet within weeks after injury. The plantar flexion is usually treated ineffectively by serial casting, and children typically develop plantarflexion contractures that require orthopedic releases. Posttraumatic spasticity often abates within 6 to 12 months, but by then, contractures have developed. The injection of BtxA into the affected muscles early after injury, as soon as significant spasticity is detected, should minimize such contractures, particularly if combined with ankle casting.
Phenol and alcohol can be injected directly into spastic muscles, where they cause focal muscle necrosis, or at the myoneural junction. Injections are painful, cause muscle soreness for several days, and are usually performed in children under a brief anesthetic. IM injections of 50% alcohol, 5 mL at one to three sites, or perineural injections of 5% aqueous phenol are common. Weakness of the injected muscle develops over several days and persists for weeks to months. Because of the pain and dysesthesias associated with their use and the variable responses obtained, phenol and alcohol are used much less frequently since the availability of Botox. However, because alcohol and phenol are far less expensive than Botox, and their effects persist longer, they are preferred in some centers to treat spastic muscles innervated by motor nerves, and they remain the injectable option of choice in developing countries.
Intrathecal Medications
Intrathecal baclofen (ITB) is by far the most common medication to be infused intrathecally, although morphine, fentanyl, tizanidine, and clonazepam have been used.
ITB is more effective than oral baclofen because the CSF concentration of baclofen obtained after intrathecal injection is so much higher than that after oral administration (400 ng/mL in a patient receiving 396 μg of ITB per day versus less than 12 ng/mL in a patient receiving 60 mg of oral baclofen per day).23 After a single ITB dose, spasticity in the lower extremities begins to diminish in 2 hours, is maximally reduced in 4 hours, and recurs in 8 to 10 hours.37 The half-life of an intrathecal dose is 4 to 5 hours.38 ITB is cleared at a rate of 30 mL/h, the rate of CSF clearance. After baclofen infusions in the region of the conus medullaris, lumbar CSF concentrations are approximately fourfold higher than those at the cisterna magna.39 These data suggest that the position of the catheter tip should vary according to whether spastic diplegia or quadriplegia is being treated.
ITB is effective in treating spasticity of spinal or cerebral origin. It diminishes stretch reflex excitability and abnormal muscle co-contractions that occur in children with cerebral spasticity.40,41 ITB has been used to treat children and adolescents with spinal spasticity due to trauma, transverse myelitis, arteriovenous malformations, or familial spastic paraparesis.42
We conducted a prospective study of long-term ITB in 37 children with cerebral spasticity that was mostly due to CP but also due to head injury or ischemia.43 We found that ITB reduced spasticity in the lower and upper extremities and that improvement occurred in hamstring range of motion and in position transitions. Improvement was also noted on an activities-of-daily-living scale that assessed communication, dressing, positioning, and feeding. Since then, multiple studies have confirmed the effectiveness of ITB in reducing spasticity, in improving motor function as evaluated by the Gross Motor Function Measure (GMFM), and in improving comfort and caregiving.44–46
Until recently, a test dose of ITB was always given to confirm the child’s response to ITB before a pump was implanted. As hundreds of children have been tested with bolus injections, it has become apparent that almost all respond to the bolus doses if they have spasticity (but not dystonia or contractures). Additional reasons not to do a test dose are that the customary doses, 50 to 100 μg, are excessive and result in extreme hypotonia of the lower extremities, in addition to the headache and vomiting that commonly occur after lumbar puncture. Thus, increasing numbers of centers that have experience with bolus doses and patient selection are forgoing the test injections and proceeding directly to pump implantation.
In children with spasticity and ventriculomegaly who may need a pump, it is important to consider a lumbar puncture to measure the opening pressure before a pump is inserted. Many children with spastic CP have had intraventricular hemorrhages as infants and have chronic ventriculomegaly and increased opening pressures.47 If such pressure is not addressed, it increases the risk for CSF leaks around the intrathecal catheter if a pump is subsequently implanted.
ITB is usually administered by a programmable pump implanted subcutaneously or subfascially in the abdomen and connected to a catheter that is tunneled subcutaneously posteriorly to the lumbar region, then via a Tuohy needle into the CSF. Nonprogrammable pumps are available but do not allow the frequent dose adjustments that are needed in the first 1 to 2 years when cerebral spasticity is being treated. Pumps are programmed to deliver doses of baclofen that are adjusted according to the goals of treatment. SynchroMed II pumps (Medtronic, Minneapolis, MN) have 20- or 40-mL reservoirs. The 20-mL pump is small enough to be inserted into children weighing 10 kg. When pumps are implanted into small, thin children, healing appears to be better if the pumps are implanted below the abdominal fascia.
The main risks of pump implantation are infection, CSF leakage, and catheter problems, such as breaking and cracking.48 The risk for each is approximately 5 to 10%. Borowski et al reported a 31% complication rate requiring surgical management over a 3-year period in 316 surgical procedures.49 If a pump infection occurs, some neurosurgeons have attempted to “save” the system by irrigating or debriding the wound and giving intravenous antibiotics for 2 to 3 weeks. In my experience, the likelihood of success with that technique is small.
ITB doses typically increase during the first year after implantation, then remain relatively stable. If a child has had good results from ITB for 1 to 2 years and then the effects wane despite increasing doses, there is almost always a problem with the catheter infusion system, not the pump. The current Medtronic pump battery lasts approximately 7 years before replacement is necessary. After a child has received ITB for several months, it should not be withdrawn abruptly. Withdrawal symptoms of itching, agitation, and spasms are common; seizures and fever may occur; and severe symptoms, such as hyperthermia, psychosis, and multiple-organ failure, have been reported.50,51 Children presenting with signs and symptoms of ITB withdrawal must be given large doses of oral baclofen, evaluated urgently, and have the malfunction repaired promptly.52
Side effects of ITB include listlessness, hypotonia of the trunk and neck, and occasional urinary hesitancy, symptoms that subside with reduction in dosage. ITB does not appear to increase the frequency of seizures. In a multicenter study of ITB in 68 children, seizure frequency was increased in none of the children.53 Overdoses, which are rare during routine treatment, cause profound muscle hypotonia and lethargy. Treatment of severe overdoses includes artificial ventilation, stopping the pump, and CSF barbotage. The drug is not toxic, and ITB infusion usually resumes after the overdose clears in 1 to 2 days.
Advantages of ITB therapy are that it is highly effective in reducing spasticity in the arms and legs and often improves global function. Furthermore, the dose can be titrated to the obtain the desired reduction in spasticity, and ITB is nondestructive. Its disadvantages are the costs of the pump, implantation, and refills, and the complications of the infusion system.
Rhizotomies
Ventral (motor) rhizotomies were described for adults with spasticity in 1932, but they fell out of favor because of muscle atrophy and the resultant loss of function.54 They have no place in the current treatment of pediatric spasticity. The neurophysiologist Sherrington55 laid the foundation for dorsal rhizotomies. He treated the “spasticity” that resulted after sectioning the midbrain of cats with rhizotomies of the cervical dorsal roots and found that their hypertonia was abolished. Parenthetically, they probably had extensor rigidity rather than spasticity. Based on that laboratory work, Foerster56 performed L2–S1 rhizotomies, sparing L4, in 153 children with cerebral spasticity and reported excellent results. The procedure was not used thereafter for several decades, probably because of the sensory loss and ataxia that would occur if the entire sensory roots of L2, L3, L5, and S1 were divided.
In 1978, Fasano et al57 reported their use of selective dorsal rhizotomy (SDR). They stimulated the lumbar posterior roots of children with CP at various rates (1 to 50 Hz) and found that the stimulation of “normal” roots at 50 Hz caused a brief muscle contraction followed by relaxation, but that stimulation of “abnormal” roots caused a tetanic contraction that often persisted after the stimulus ceased. They divided the rootlets that generated the “abnormal” response and reported good relief of lower extremity spasticity in 71% of cases, with return of spasticity in approximately 5% of cases in the following years. This procedure was modified and popularized by Peacock et al, who identified as “abnormal” features tetanic responses, responses that persisted after the stimulus ceased, clonus, contraction of muscles in the contralateral extremity, and contraction of muscles not normally in that myotome.1 They also performed the testing and division of the nerves at their exit zones from the dural sac, where they could be identified more readily than at the point where they entered the conus medullaris. Park and Johnston subsequently advocated dorsal rhizotomies at the conus, via limited laminotomies, and reported equally good outcomes.58 Ou et al compared outcomes after single-level laminotomies at the conus versus multilevel lumbar laminectomies and found no differences in outcomes other than shorter hospitalizations after the single-level procedure.59
There is continued controversy about the validity of the neurophysiologic criteria used to distinguish normal and abnormal responses. It appears that tetanic contractions, which were considered abnormal, are a normal response to a 50-Hz stimulus. Investigators have demonstrated that there is considerable variability in response patterns to repetitive stimuli.60–62 Some of the response variability may be attributable to whether stimulation is performed with constant current or constant voltage. Because tetanic responses are not considered now to be abnormal, the proportion of nerve rootlets divided has decreased (without a decrease in effectiveness), and the frequency of postoperative hypotonia has diminished.
Many neurosurgeons now perform lumbar dorsal rhizotomies without using neurophysiogic monitoring and base the percentage of dorsal roots to be divided on the preoperative clinical examination. Steinbok et al compared the outcomes of 22 patients who had lumbar dorsal rhizotomies without electrophysiologic guidance and the outcomes of 22 patients who had lumbar dorsal rhizotomies with electrophysiologic guidance, and they found no differences in spasticity reduction or any other parameters.63 I used monitoring for perhaps 15 years, but for the past 7 years I have used no monitoring and have observed no difference in spasticity outcomes.
The critical aspect of SDR is not electrical stimulation, but rather patient selection. Good candidates for SDR are usually 5 to 8 years old with spastic diplegia that is inhibiting their gait; they have good strength in their legs, and mild contractures. Few children with CP meet these criteria. Functional decline in children undergoing SDR after age 10 has recently been reported.64 Dorsal rhizotomies do not improve dystonia, although they are performed relatively often in children with unrecognized dystonia, nor do dorsal rhizotomies reliably improve spasticity in children with familial spastic paraparesis.
Children treated by Peacock’s method have significantly decreased spasticity in the lower extremities, improved range of motion, and improved gait, with longer stride length and faster cadence. McLaughlin et al65 performed a meta-analysis of data from three RCTs of SDR. Spasticity was significantly decreased, and function, as evaluated by the GMFM, was significantly improved. Although SDR has been reported to decrease the development of musculoskeletal deformities and contractures, more recent publications indicate that many children will require orthopedic operations.66,67
Although some series report a regression of the GMFM to baseline values 10 years after SDR, most studies report persistent improvement in GMFM and GMFCS levels.66–68 Two of the best long-term evaluations of SDR, from Peacock’s original group, found that 20 years after SDR, a prospective gait analysis in 13 patients showed improved locomotor function and improvements in the GMFCS levels.69,70 Spasticity, stride length, and quality of gait may also improve after SDR in children with spastic hemiparesis.71
Cervical SPR has been performed to treat spasticity of the biceps and of the wrist and finger flexors, which results in contractures and poor hygiene.72 In my experience, cervical SDR improves spasticity in the upper extremities approximately as well as lumbar SPR improves spasticity in the lower extremities, but upper extremity function is less likely to improve. Cervical SDR is infrequently performed, mainly because of the availability of treatment alternatives such as IM injections, peripheral neurectomies, and orthopedic operations.
Peripheral Neurectomies
Peripheral neurectomies have been used in Europe and India more often than in the United States and are an underutilized operation. Like IM injections, they do not reduce spasticity per se but rather weaken contraction of the muscle. Thus, they are performed to treat focal spasticity rather than generalized spasticity. Spasticity in the plantar flexors, hip adductors, and elbow flexors is treated by peripheral neurectomies of the posterior tibial, obturator, and musculocutaneous nerves, respectively.73 The procedure is performed to increase range of motion, facilitate care, and lessen the development of contractures, rarely with the goal of improving function. It is performed under general anesthesia and can be done either nonselectively, dividing 50 to 80% of the nerve, or selectively, with intraoperative stimulation of nerve fascicles to identify motor branches whose stimulation results in strong muscle contractions. Selective motor fasciculotomy for spastic upper extremities was reported for 20 patients whose mean age was 13 years.74 Fasciculotomies of the musculocutaneous, median, and ulnar nerves reduced spasticity of the affected muscles and improved self-care.
Treatment Selection
Given the plethora of methods available to decrease spasticity, both those who treat and those who receive treatment may be perplexed. It is important to remember that not all spasticity needs to be treated, and that treatment goals should be carefully identified before any therapy is undertaken. The reasons for not needing to treat all patients with spasticity include the following: (1) Some mild spasticity causes no interference with function and no orthopedic problems. (2) Some spasticity is clinically useful in individuals with weak extremities, to augment contractions of weak muscles. (3) Spasticity that causes extremity stiffness may help maintain an erect posture of the neck and trunk; if it were treated, the arms and the legs might relax, but the child might then be unable to hold the head up or to sit upright.
In general, treatment goals include improving function, facilitating care, preventing or minimizing contractures, and, infrequently, reducing painful muscle spasms or cramps. The goal of improving function may include general goals such as easier ambulation, dressing, or transfers, but unfortunately, more specific goals such as improved handwriting or swallowing cannot be predicted. In my experience, upper extremity function improves in approximately 60% of children with spastic quadriparesis after ITB, and speech intelligibility and swallowing improve in approximately 40%, but I know of no way to predict which children will have such improvement; screening trials do not answer the question.
Treatment selection also depends on the severity of the spasticity, the extent of involvement (focal or diffuse), and the child’s age. Muscle tone with a score of 2 on the Ashworth Scale is mild and usually does not need to be treated; tone with a score of 3 to 5 usually does. Oral medications are of minimal, if any, help in treating severe spasticity. Focal spasticity is more appropriately treated with botulinum toxin injections than is diffuse spasticity.
Age is important for the following reasons: (1) Ablative procedures like SPR are usually delayed until the age of 4 to 5 years because brain maturation (and a decrease in spasticity) may occur by that time. (2) Children younger than 4 years are infrequently candidates for ITB pumps; their spasticity can usually be treated satisfactorily with oral medications and botulinum toxin. (3) SPR is infrequently performed in children older than 16 years if the goal is to improve function because their ability to strengthen weak muscles and to learn new gait patterns is more limited than that of younger children. Ablative procedures are more commonly performed in children for whom the treatment goal is to facilitate care rather than to improve function.
For children 1 to 4 years old with diffuse spasticity, treatment usually entails oral spasmolytics, which usually reduce Ashworth Scale scores from 3 or 4 down to 2. However, the score is infrequently reduced to normal, and infrequently to a degree such that the child no longer exhibits plantar flexion of the feet or scissoring (adduction) of the legs; botulinum toxin is often injected into those muscles to prevent contractures that might otherwise develop despite the oral medications.
The age at which SPR is performed varies according to the treatment goal: if improved ambulation is the goal, SPR is performed most commonly in children 5 to 8 years old but is infrequently performed after age 12. If the goal is to facilitate care and age is not an issue, the operation can be done at any time, preferably before contractures develop. If the goal is to facilitate the care of children with spastic quadriparesis (whose spasticity involves the lower extremities more than the upper), lumbar SPR is probably the best treatment—it is definitive and effective. Of children with spastic quadriparesis that involves the upper and lower extremities equally, those who are functional are probably best treated by ITB and those who are nonfunctional either by ITB or by combined cervical and lumbar dorsal rhizotomies.
ITB is useful in ambulatory children who have some leg weakness because doses can be increased slowly as muscle strength improves. ITB is probably not helpful in improving the ambulation of patients with spastic diplegia who are in their late teens or older because of their limited ability to improve their strength and learn new gait patterns, although it may improve trunk and upper extremity mobility and function in that age group. ITB is also appropriate for children with spastic paraparesis after spinal cord injuries. For those with nonfamilial spastic paraplegia, either SPR or ITB may be used; SPR is probably preferable because of its lower complication rate. Children with spastic quadriparesis after spinal cord injuries are usually treated with ITB.
Peripheral neurectomies may be considered in children with focal spasticity that has not yet caused substantial contractures. The children have usually been given repeated botulinum toxin injections for 1 to 3 years but need therapy that is more permanent.
76.2 Dystonia
Dystonia has been defined as “… a movement disorder in which involuntary sustained or intermittent muscle contractions cause twisting and repetitive movements, abnormal postures, or both.”2 Dystonia is classified according to the body regions involved: focal if one region (such as one upper extremity) is involved, segmental if adjacent regions (such as the neck and upper extremity) are involved, hemidystonia if one side of the body is affected, and generalized if the entire body is affected. Dystonia is also characterized as primary or secondary: primary if no underlying pathology can be identified and the individual shows no other neurologic abnormalities, and secondary if the dystonia can be attributed to a structural cause (such as a lesion of the basal ganglia). Some clinicians add a third category, heredodegenerative. The term primary dystonia is synonymous with the previously used term, dystonia musculorum deformans. Whether primary or secondary, dystonia is characterized by simultaneous contractions of agonist and antagonist muscles, and to a considerably greater extent than occurs in spasticity.
Primary dystonias are diagnosed by genetic analysis. At present, 21 different abnormalities in the DYT (TORIA) gene have been identified, causing disorders such as early-onset primary dystonia (DYT1), X-linked dystonia–parkinsonism (“Lubag,” DYT3), dopa-responsive dystonia (Segawa syndrome, DYT5), and myoclonic dystonia (DYT11). Secondary dystonias are diagnosed most often by abnormalities in the basal ganglia, particularly the putamen. Heredodegenerative dystonias may be caused by Huntington disease, Wilson disease, neuroacanthocytosis, Rett syndrome, Leigh disease, and neurodegeneration associated with iron deposition (Hallervorden-Spatz disease, PKAN [pantothenate kinase–associated neurodegeneration] disease), and are diagnosed by tests specific for each of those disorders.
Pediatric neurosurgeons encounter dystonia frequently in children with CP, occasionally after head injury, stroke, or tumor removals; uncommonly in children with heredodegenerative disorders; and rarely in children with primary dystonia. CP usually causes generalized dystonia but occasionally hemidystonia or dystonic paraparesis, whereas stroke or head injuries are more likely to cause focal dystonia or hemidystonia and to follow an initial hemiplegia. In the series of Saint Hilaire et al,75 no patient with CP had focal dystonia. Posttraumatic dystonia usually occurs after severe head injuries but may follow minor injuries, and in either case it may develop months to years after the injury and may worsen for a few years before stabilizing.76 As a general rule, dystonia that develops several years after an injury or an ischemic event is less likely to be severely disabling than dystonia that begins soon afterward.
76.2.1 Epidemiology
Primary dystonia occurs most frequently in Ashkenazi Jews, affecting 1 per 3,000 to 1 per 9,000 in that population. In non-Jewish people, the incidence ranges between 1 per 10,000 and 1 per 30,000. Secondary dystonia is far more common, occurring in 15 to 25% of individuals with CP.
76.2.2 Clinical Presentation
Dystonia associated with CP may not be clinically evident until 3 to 15 years of age, perhaps because a certain degree of myelination is necessary before the dystonic movements can become apparent.77–79 Dystonic movement in CP may cause the extremities to writhe in random motions or to contract repeatedly in abnormal postures. Dystonia may cause intermittent deviation of the head to one side, posterior arching of the neck and trunk, and intermittent extension or adduction of the upper and lower extremities. The movements are not stereotypic, as are the abnormal postures seen in infancy (e.g., the asymmetric tonic neck reflex). Dystonia is increased by excitement and ceases with sleep. Dystonic children are typically thin because of the calories expended during sustained muscle contractions, and they have muscles that are well developed because of repeated isotonic contractions. Although the brain lesions in CP are static, the dystonic movements may worsen over several years.
Primary dystonia in children is graded most frequently with the Burke-Fahn-Marsden (BFM) Dystonia Rating Scale, which was developed to grade primary dystonia in adults.80 Secondary dystonia is graded with either the Barry-Albright Dystonic (BAD) Scale, which was developed and validated for use in children with secondary dystonia, or with the BFM Scale.81
Dopa-responsive dystonia (Segawa disease) may be misdiagnosed as dystonic CP.82 Dopa-responsive dystonia begins during childhood, often with gait difficulties or as dystonia in a foot. It has diurnal variations in 75% of cases and frequently a positive family history, and it may be associated with spasticity or Parkinsonian features. Dopa-responsive dystonia responds dramatically and chronically to low doses of levodopa, without the dyskinesia that occurs in adults treated with levodopa.83 A trial of levodopa should be considered in children with dystonia unless they have an unequivocal historical and radiographic features of CP.
76.2.3 Pathology and Pathophysiology
There is no demonstrable pathology in primary dystonia. Secondary dystonias and some heredodegenerative dystonias are associated with lesions in the basal ganglia, particularly in the caudate and putamen (▶ Fig. 76.2 and ▶ Fig. 76.3).
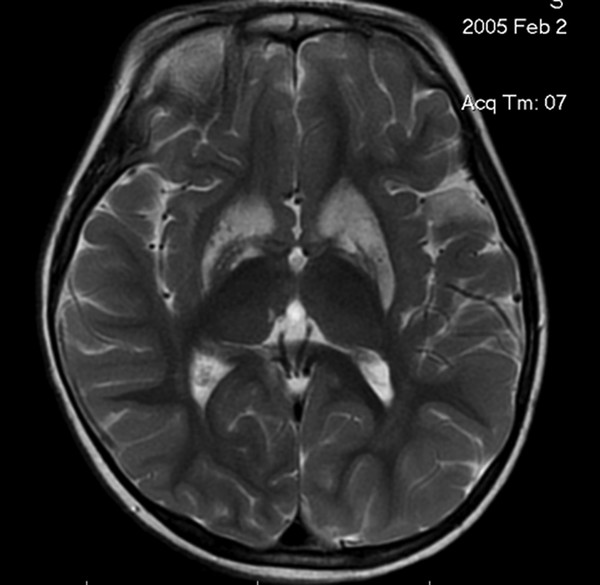
Fig. 76.2 Axial magnetic resonance image demonstrating caudate and putaminal changes associated with generalized secondary dystonia.









