Chapter 50 Spasticity
Classification, Diagnosis, and Management
• Spasticity is velocity-dependent resistance to passive muscle stretch that usually originates from pathological issues with the brain or spinal cord. Symptoms of spasticity include muscle tightness, cramping/pain, and fatigue. Spasticity is primarily due to an imbalance between activation and inhibition of muscle groups.
• Cerebral palsy is the most common cause of spasticity, especially in children. Stroke, multiple sclerosis, and brain or spinal trauma are common causes of spasticity in adults.
• Spasticity results from decreased inhibition of muscle groups and facilitation of hypertonia. The decreased inhibitory input into the motor unit results in coactivation of both agonistic and antagonistic muscle groups during volitional movement, and creates an imbalance of excitatory neurotransmitters in the spinal cord, specifically a lack of the inhibitory substance γ-aminobutyric acid (GABA).
• The Ashworth or Modified Ashworth scales are commonly used to classify the severity of spasticity. The Modified Ashworth scale1 ranges from 0 (no increase in muscle tone) to 4, in which affected part(s) are rigid in flexion or extension. Management of spasticity is often achieved with a combination of medications and surgical procedures.
• Intrathecal baclofen (ITB) pumps are most effective in pediatric patients older than 3 years when a response to a baclofen “test dose” is observed. ITB is effective in patients with spasticity in all limbs and in dystonia. Baclofen activates GABAB receptors, and benzodiazepines activate GABAA receptors; both provide compensatory inhibition in the brain and spinal cord.2 Spastic patients with intrathecal or oral balcofen withdrawal present with increased tone, pruritus, and anxiety. Severe withdrawal may include fever, seizures, hallucinations, and rarely death. Treatment of withdrawal includes administration of oral baclofen or intravenous benzodiazepines.
• Botulinum toxin injection is useful in treating spasticity of individual muscle groups or extremities.
• Selective dorsal rhizotomy is most effective in the treatment of spastic diplegia, and is more effective than physical therapy alone in selected patients. Cost-effectiveness studies have shown an advantage of selective dorsal rhizotomy (SDR) over ITB in patients with spastic diplegia due to reduced hospital readmissions for pump replacements and complications.
Spasticity is defined as a “velocity-dependent, increased resistance to passive muscle stretch.”3 It is distinguished from other common hypertonic movement disorders (such as dystonia) by quantifying the amount of abnormal movement: Spasticity is isokinetic (abnormal but not increased movements), while dystonia and other movement disorders are hyperkinetic (abnormal and increased movements).4 It is often associated with muscle tightness, cramping/pain, and fatigue, and its severity may range from mild to disabling. Even though spasticity is considered a diagnosis unto itself, it is always related to an underlying injury to the brain or spinal cord. The treatment of spasticity involves a combination of medical and surgical management.
Causes
Spasticity is typically defined by the causative diagnosis. The most common causes are cerebral palsy (CP),5 traumatic injury (including injuries to either the brain6,7 or spine8), stroke,9 and multiple sclerosis (MS);10 other inherited disorders (e.g., Wilson’s disease, Hallervorden-Spatz disease) are rare causes.
Cerebral Palsy
Cerebral palsy is the most commonly encountered spastic disorder, occurring in 2.4 per 1000 children and accounting for up to 75% of affected patients.11,12 Cerebral palsy is usually described as “a range of nonprogressive syndromes of posture and motor impairment that results from an insult to the developing central nervous system.”13 One or both of the following signs are required for diagnosis, according to the task force for pediatric hypertonia: (1) an increase in resistance to externally applied movement in the same direction as joint movement; and (2) a rapid rise in resistance to externally applied movement above a threshold speed or joint angle.5
Brain or Spinal Trauma
Injuries to either the brain or spinal cord can result in spasticity.6,7,14,15 These injuries include involvement of any of the following: basal ganglia, cerebellum, motor cortices, and spinal cord. It is estimated that up to 25% of patients with moderate or severe trauma develop spasticity symptoms.7
Stroke
Similar to traumatic spasticity, stroke-related spasticity is commonly encountered after lesions to either upper motor neurons or descending inhibitory systems (see later discussion). Approximately 15% to 40% of patients develop clinical spasticity disorders after major stroke.9,16,17
Multiple Sclerosis
Multiple sclerosis is a demyelinating disease often affecting upper motor neuron areas of the brain and spinal cord. A substantial proportion of MS patients develop spastic symptoms; one major study found that 84% of a large database of MS patients suffered from at least minimal spasticity during the disease course, with 34% reporting moderate to severe or disabling symptoms.10
Classification
Many practitioners further classify spasticity within the following anatomical categories, first described for CP:18
 Quadriparesis/tetraplegia—involving all four extremities
Quadriparesis/tetraplegia—involving all four extremities
 Paraparesis/diplegia—involving the bilateral lower extremities
Paraparesis/diplegia—involving the bilateral lower extremities
 Hemiparesis—involving ipsilateral upper and lower extremities
Hemiparesis—involving ipsilateral upper and lower extremities
 Monoparesis—involving one extremity
Monoparesis—involving one extremity

Pathophysiology
Understanding the neurophysiology of spasticity requires detailed knowledge of the control and regulation of movement and posture in the central nervous system. In normal conditions, a balance exists between alpha motor neurons in the spinal cord (activated by descending corticospinal tracts) and the descending inhibition created by the basal ganglia, cerebellum, and reticular activating system. These inhibitory systems exert their control on the spinal cord via descending input from the reticulospinal and other tracts, which synapse on the motor neurons or interneurons of the spinal cord. This system maintains baseline muscle tone and posture, as well as facilitates the complementary deactivation of antagonistic muscle groups via Ia and Ib afferents in the dorsal root of the motor unit. For example, flexion of the thigh is facilitated by positive input from the corticospinal tracts in conjunction with deactivation of extensors via descending inhibitory tracts.19,20
Spasticity results from decreased inhibition or facilitation of hypertonia.19,20 The decreased inhibitory input into the motor unit results in coactivation of both agonistic and antagonistic muscle groups during volitional movement, and creates an imbalance of excitatory neurotransmitters in the spinal cord (in particular, a lack of the inhibitory substance γ-aminobutyric acid [GABA]). Chronic spasticity may lead to denervation supersensitivity, and ultimately permanent soft tissue contractures requiring surgical release, especially in the ankle joint and Achilles tendon.21
Diagnosis
A careful history and physical examination is imperative for the diagnosis of both the cause and classification of spasticity. In pediatric patients with CP-related spasticity, a failure to meet motor system and other developmental milestones is often apparent by age 1 to 2 years, and is most severe by age 3.13 Often, symptoms of CP will decline over childhood, with one major study describing 66% of patients with spastic diplegia and 50% with CP of any kind “outgrowing” their symptoms by age 7.22 Older patients with alternative causes of spastic disorder will complain of characteristic muscle tightness, cramping, pain in the affected extremities, and generalized as well as focal fatigue.
Physical examination findings are often linked with the location of injury. Patients with CP often manifest with di- or tetraplegia of varying severity, often with disproportionate involvement of flexors, adductors, and internal rotators.4 In patients with cervical spinal cord injury, both flexor muscles in the arms and extensors in the leg (so-called antigravity muscles) are often affected, but lesions in the thoracic or lumbar spine typically affect only leg extensors. Cortical spasticity from stroke or trauma is often related to the laterality of injury, with contralateral extremities or a single extremity affected.23 Multiple sclerosis patients are heterogeneous in presentation depending on the areas of demyelination, but often present with leg adductor and extensor imbalance.
Resistance to passive muscle stretch correlating with the speed of the stretch is the hallmark of the physical examination in affected muscle groups in patients with spasticity. Several clinical scales have been developed to determine the severity of symptoms. The Ashworth scale grades spasticity as follows: 0 = normal muscle tone; 1 = slight increase in muscle tone, “catch” when limb moved; 2 = more marked increase in muscle tone, but limb easily flexed; 3 = considerable increase in muscle tone; and 4 = limb rigid in flexion or extension.24 The Modified Ashworth scale1 provides further precision, especially in patients with hemiplegia: 0 = no increase in muscle tone; 1 = slight increase in muscle tone, manifested by a catch and release or by minimal resistance at the end of the range of motion (ROM); 1+ = slight increase in muscle tone, manifested by a catch, followed by minimal resistance throughout the remainder (less than half) of the ROM; 2 = more marked increase in muscle tone through most of the ROM, but affected part(s) easily moved; 3 = considerable increase in muscle tone, passive movement difficult; 4 = affected part(s) rigid in flexion or extension. Most studies of the diagnosis and treatment of spasticity employ these scales, as well as other indices of motor function and dexterity, to grade the physical findings of individual patients.
Treatment
The treatment of spasticity almost always involves multidimensional or multimodality therapy that may include physiotherapy, medical therapy (oral medications, percutaneous injections), neurosurgical therapy (surgical implantation of intrathecal infusion pumps, permanent selective denervation procedures such as selective dorsal rhizotomy), and orthopedic procedures.25
Medical Therapy
The most common oral medications used in the treatment of spasticity include baclofen, benzodiazepines, tizanidine, dantrolene, gabapentin, and pregabalin. A summary of these medications is found in Table 50.1. They rely on two common mechanisms to reduce spastic symptoms throughout the body, often in combination: (1) compensating for reduced GABA or other inhibitory neurotransmitters in the brain or spinal cord, or (2) reducing the amount of excitatory neurotransmitters through direct inhibition or activation of inhibitory interneurons. The common mechanism of action of these medications also results in a similar side effect profile, including most commonly somnolence, ataxia, and muscle weakness.15,26
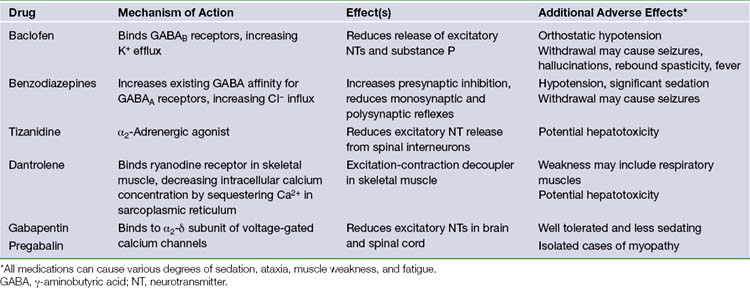
Baclofen, the most common oral medication in the treatment of spasticity, binds to GABAB receptors in Rexed laminae I to IV of the spinal cord.27 Activation of these metabotropic receptors leads to increased cell permeability of K+, causing cation efflux and resultant hyperpolarization of the cell membrane.12 This hyperpolarization leads to a reduction in the release of excitatory neurotransmitters (such as glutamate and aspartate), as well as substance P. Important side effects of baclofen include orthostatic hypotension and withdrawal symptoms such as seizures, fever, hallucinations, and rebound spasticity. One drawback of oral baclofen is poor penetration of the blood-brain barrier with cerebrospinal fluid (CSF) drug levels 10-fold lower than serum concentrations. It is common that oral dose escalation leads to side effects before full therapeutic efficacy is reached. Intrathecal delivery of baclofen is highly effective and is the favored modality of drug delivery when patients are unable to tolerate increased doses of oral baclofen (see later discussion).
Benzodiazepines facilitate the binding of existing GABA to the GABAA receptor,28 found in high concentrations in the reticular formation and polysynaptic spinal tracts. In contrast to the GABAB receptor, the GABAA receptor is ionotropic. Its activation results in the increase in cell permeability of Cl−, causing anion influx and hyperpolarization. This hyperpolarization increases presynaptic inhibition and reduces monosynaptic and polysynaptic reflexes throughout the spinal cord. A variety of different benzodiazepine compounds are available including oral, intravenous, and rectal routes of administration, with different durations due to the rate of metabolism. The most important (and often dose-limiting) side effect of this medication class is sedation. Rapid reversal of benzodiazepine toxicity can be achieved with the administration of flumazenil. Additionally, habituation and tolerance may develop, and prolonged use is associated with addiction.
Tizanidine and related medications (such as clonidine) provide an alternative mechanism to the preceding medications via the reduction of excitatory neurotransmitters. Tizanidine is an α2-adrenergic agonist that acts throughout the central nervous system (CNS) to reduce excitatory amino acid release from spinal interneurons.29,30 Tizanidine is particularly effective in adult spasticity from spinal disease or multiple sclerosis.31 Important adverse effects include nausea and vomiting, hypotension (clonidine), and sedation (tizanidine).
Dantrolene is unique among oral medications in the treatment of spasticity due to its effects on skeletal muscle. Dantrolene binds to skeletal muscle ryanodine receptors, preventing the release of calcium into the cytosol from its sequestration in the sarcoplasmic reticulum during motor unit activation. Thus, its action is defined as an “excitation-contraction decoupler” in skeletal muscle.32–34 It is also used in the emergency treatment of malignant hyperthermia and neuroleptic malignant syndrome. Although it is less sedative than other antispasticity agents, its effect on skeletal muscle may result in significant weakness of both volitional and (in severe cases) respiratory muscles.35 Another important side effect is the approximately 1.8% risk of hepatotoxicity, which is increased with coadministration of other agents (i.e., valproate, tizanidine).36 Some clinicians recommend routine surveillance of liver function tests in patients taking dantrolene.
Two newer medications, gabapentin and pregabalin, were originally developed as anticonvulsants but have found utility in the management of spasm and other disorders such as neuropathic pain. Both medications work in a similar fashion as GABA analogs that bind to the α2δ-subunit of voltage-gated calcium channels, inhibiting calcium influx. This mechanism reduces the concentration of excitatory neurotransmitters such as glutamate and aspartate in the CNS.37,38 Gabapentin is a well-established therapy for the treatment of spasticity of spinal origin39,40 and in the management of MS.41 Pregabalin is emerging as an alternative agent with similar properties.42 Both medications are well tolerated with minimal side effects, such as sedation, even after significant titration. Gabapentin has been associated with myopathy, especially in patients with renal failure, as it is renally excreted,43 and pregabalin is somewhat more sedating and may cause dose-limiting ataxia.44
Recent advances have been made in the treatment of spasticity of focal hypertonia using percutaneous injection of botulinum neurotoxin (BoNT), often in conjunction with the general effects of oral medications or surgical treatments,45 or as monotherapy for isolated limb spasticity.46–48 BoNT is derived from Clostridium botulinum exotoxin, and acts by cleaving polypeptides required for exocytosis, such as synaptosomal-associated protein (SNAP)-25, vesicle-associated membrane protein (VAMP), and syntaxin, within the cytosol of motor neurons.49,50 Cleavage of these peptides prevents the release of acetylcholine into the neuromuscular junction, relieving focal muscle hypertonia. Additional indirect beneficial effects on muscle tone, such as the suppression of other excitatory neurotransmitters and reduced autonomic activity, may occur. A single treatment remains effective for 2 to 4 months at a time,26 providing a useful adjunct treatment for persistent symptoms involving a single muscle group, such as the calf, upper extremity, leg adductor, or cervical musculature.
There are three main adverse effects of BoNT injection.45 First, inhibition of acetylcholine release may spread to neighboring nerve endings, including respiratory muscles. This has led to a black-box warning for all BoNT formulations regarding the risk of potentially life-threatening respiratory failure. Second, sustained or repeated administration to a single anatomical area may produce effects similar to denervation, such as muscle atrophy. Third, the patient’s host response may generate antibodies to the BoNT protein, leading to immunoresistance that prevents its association with neuronal membranes.51 These antibodies may lead to a habituation to BoNT injections over time.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


