Chapter 5 Spinal Dysraphism and Tethered Spinal Cord
• Spinal dysraphism refers to anomalies of the spine in which the midline structures do not fuse. Myelomeningocele is the most common significant birth defect involving the spine.
• The prevalence of spina bifida in industrialized countries has been decreasing because of the steadily increasing proportion of affected fetuses that are detected prenatally and electively terminated. In addition, there is strong scientific evidence that the use of preconception folate appears to decrease the risk of developing a neural tube defect such as myelomeningocele.
• Embryologically, the abnormality manifests between 3 and 4 weeks of gestation, during the period called neurulation. The abnormality represents the failure of the posterior neuropore to close properly. Patients with myelomeningocele usually have hydrocephalus and a Chiari II malformation. Surgical closure of the dorsal defect is performed shortly after birth.
• In utero closure of myelomeningocele is a promising surgical technique that has been pioneered at several medical centers. The goal is to decrease the incidence of hydrocephalus and hindbrain abnormalities found in this population. A randomized clinical trial demonstrated efficacy of this treatment option in reducing shunt placement and improving motor function.
• Developmental anomalies involving the caudal portion of the neural tube are increasingly important in clinical practice. This is the result of advances in radiological diagnostic techniques and a consequent change in the philosophy of treatment, which includes prophylactic cord untethering to prevent neurological deficits. Greater awareness of the conditions of lipomyelomeningocele, tethered cord, diastematomyelia, and sinus tracts, by pediatricians, orthopedists, pediatric surgeons, and urologists, in concert with the widespread application of magnetic resonance imaging in addition to the clinical examination, have led to earlier recognition of these congenital surgically correctable problems. Recognition of the cutaneous, orthopedic, neurological, and urological stigmata of “occult” spinal anomalies has been helpful for early diagnosis.
• Patients with sacral agenesis, cloacal extrophy, and other caudal regressions syndromes may require magnetic resonance imaging after initial pediatric surgery intervention to identify potential tethered cord anatomy.
Myelomeningocele
Myelomeningocele is the most common significant birth defect involving the spine. Since the early 1980s the prevalence of spina bifida in industrialized countries has been decreasing because of the steadily increasing proportion of affected fetuses that are detected prenatally and electively terminated. The incidence of the condition ranges from less than 1 case per 1000 live births in the United States to almost 9 cases per 1000 in areas of Ireland. The etiology is unknown, but evidence exists for both environmental and genetic influences. A role for genetic risk factors is supported by numerous studies documenting familial aggregation of this condition. In addition, several lines of evidence point to the potential importance of maternal nutritional status as a determinant of the risk for having a child with spina bifida. Indirect evidence for this association is provided by studies indicating that the season of conception, socioeconomic status, and degree of urbanization may be related to the risk of spina bifida. In August 1991, the Centers for Disease Control and Prevention (CDC) advised that women with a history of an affected pregnancy should take 4 mg of folic acid daily, starting at the time they planned to become pregnant, after publication of the Medical Research Council in Britain Vitamin Study Group report.1 This recommendation was based on a randomized, double-blind, multicenter study performed in Europe that clearly showed the protective effect of periconception folate in reducing the recurrence of spina bifida when ingested by the mothers who had previous births of children with spina bifida. A second randomized, double-blind study was performed in Hungary and demonstrated conclusively the beneficial effects of periconception folic acid intake by mothers on decreasing the incidence of first occurrence of spina bifida.2 It was anticipated that these recommendations would have a substantial impact on reducing the risk of neural tube defects in the offspring of such women. Although it is hoped that this benefit will be the case, it should be noted that the vast majority of affected pregnancies (approximately 95%) occur in women with no history of a prior affected fetus or child.3 Currently the recommended daily dose of folic acid is 0.4 mg for all women of childbearing age who are capable of becoming pregnant. Fortification of the food supply may be a more effective strategy at preventing neural tube defects, rather than individual supplementation.
Recent developments in the prenatal diagnosis of fetal anomalies have made antenatal recognition of myelomeningocele commonplace. Families at risk are routinely offered amniocentesis for amniotic alpha fetoprotein and acetylcholinesterase, which are important in separating open lesions from skin-covered masses, such as myelocystocele. Amniocentesis along with ultrasound screening has a combined accuracy of more than 90%. Prenatal magnetic resonance imaging (MRI), using ultrafast T2-weighted sequences, may also be used to characterize the Chiari II malformation and other associated anomalies.4 Furthermore, fetal MRI may augment ultrasound by detecting spinal cord abnormalities underlying bony abnormalities.5 Recent studies indicate that such prenatal imaging studies can help to determine prognosis. Specifically, lesion level determined by prenatal imaging studies appears to predict neurological deficit and ambulatory potential, but not the degree of fetal ventriculomegaly or the extent of hindbrain deformity.6 Families can be professionally counseled regarding the expected prognosis and decisions about abortion or the new option of fetal closure.
The majority of fetuses with spina bifida that are not electively terminated receive no specific treatment until after birth. In the United States, these babies are generally delivered by cesarean section.7 However, the benefit of this approach relative to vaginal delivery has not been clearly demonstrated. Data suggest that if broad-spectrum antibiotics are administered, closure of the myelomeningocele can be safely delayed for up to a week to allow time for discussion with the parents. In most instances, however, the closure is performed within 48 to 72 hours of birth. The parents should be told the infant’s prognosis based on the functional spinal level, and it should be emphasized that closure of the defect is a life-saving measure but will not alter the preexisting neurological deficits. Pending plans for definitive care, the infant is nursed in the prone position with a sterile, saline-soaked gauze dressing loosely applied over the sac or neural placode.
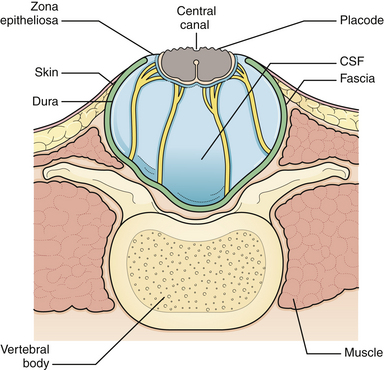
The initial step in managing the newborn with myelomeningocele is a careful physical examination by a pediatrician and neurosurgeon. A thorough evaluation should reveal associated anomalies, including cardiac and renal defects that might contraindicate surgical closure of the spine defect. Approximately 85% of myelomeningocele patients either present with hydrocephalus or develop it during the newborn period.8 A large head or bulging fontanelle suggests active hydrocephalus and indicates the need for a head ultrasound or computed tomography (CT) scan. Stridor, apnea, or bradycardia in the absence of overt intracranial hypertension suggests a symptomatic Chiari II malformation, which carries a poor prognosis. The myelomeningocele is inspected; the red, granular neural placode is surrounded by a pearly “zona epitheliosa” that must be entirely excised to prevent the appearance of a dermoid inclusion cyst. Most myelomeningoceles are slightly oval with the long axis oriented vertically. If the lesion is oriented more horizontally, a horizontal skin closure may be preferable. Neurological examination is difficult in a newborn infant, and it is hard to separate voluntary leg motion from reflex movement. It must be assumed that any leg movement in response to a painful stimulus to that limb is reflexive. Contractures and foot deformity denote paralysis at that segmental level. Virtually all affected neonates have abnormal bladder function, but this is difficult to assess in the newborn. A patulous anus lacking in sensation confirms sacral denervation.
Generally, the back is closed first, and a CSF shunting procedure is deferred unless necessary. In cases with overt hydrocephalus, the back closure and the shunt can be performed at the same time to protect the back closure from CSF leakage. The goal of back closure is to seal the spinal cord with multiple tissue layers to inhibit the entrance of bacteria from the skin and to prevent CSF leakage while preserving neurological function and preventing tethering of the spinal cord. Accomplishing this goal requires a thorough understanding of the three-dimensional anatomy of the tissue layers involved (Fig. 5.1).
Surgical Technique
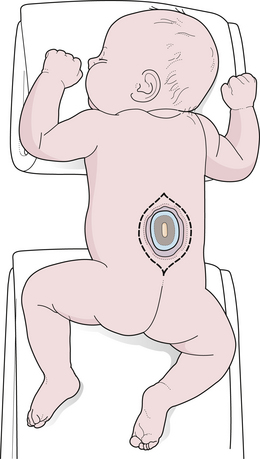
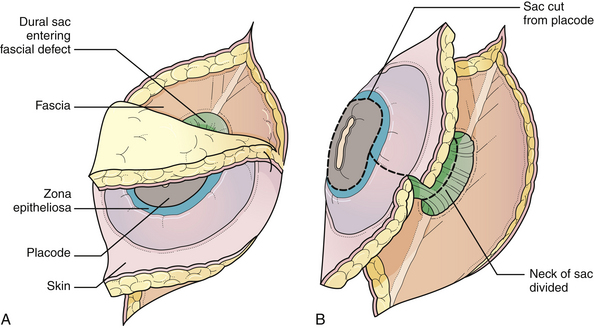
General anesthesia is used, and the patient is placed in the prone position, with rolls under the chest and hips to allow the abdomen to hang freely (Fig. 5.2). If the sac remains intact, fluid is aspirated and sent for culture. The surgeon gently attempts to approximate the base of the sac or defect vertically, then horizontally, to determine which direction of closure will produce the smallest skin defect. An elliptical incision is made, oriented along that axis, just outside the junction of the normal, full-thickness skin and the thin, pearly zona epitheliosa. Full-thickness skin forming the base of the sac is viable and should not be excised. The incision is carried through the subcutaneous tissue until the glistening layer of everted dura or fascia is encountered. The base of the sac is mobilized medially until it is seen to enter the fascial defect (Fig. 5.3A). The sac is entered by radially incising the cuff of skin surrounding the neural placode. This skin is sharply excised circumferentially around the placode with scissors and discarded (Fig. 5.3B). It is important to excise all of the zona epitheliosa to prevent later formation of an epidermoid cyst. At this point, the neural placode is lying freely above the everted dura (Fig. 5.4).
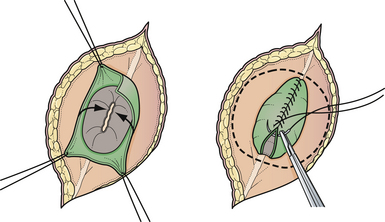
Attention is then directed to the dura, which is everted and loosely attached to the underlying fascia. The dura is undermined bluntly and reflected medially on each side until enough has been mobilized to enable a closure (see Fig. 5.4). The dura is closed in a watertight fashion using a running suture of 4-0 neurilon. If possible, the fascia is closed as a separate layer by incising it laterally in a semicircle on both sides, elevating it from the underlying muscle, and reflecting it medially. Like the dura, the fascia is closed with a continuous stitch of 4-0 suture material (Fig. 5.5). The fascia is poor at the caudal end of a lumbar myelomeningocele as well as with most sacral lesions; thus, closure at this level may be incomplete.
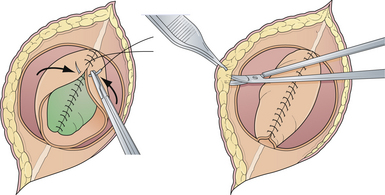
The skin is mobilized by blunt dissection with dissecting scissors or a finger. It may be necessary to free up the skin ventrally all the way to the abdomen (see Fig. 5.5). In most instances, midsagittal (vertical) plane closure is easiest, but occasionally horizontal closure results in less tension. A two-layer closure using vertical interrupted mattress skin sutures is preferred.
Very large lesions require special techniques. Various types of “Z-plasties” and relaxation incisions have been described and may be necessary in very large or difficult lesions. Large circular defects can be closed using a simple rotation flap (Fig. 5.6). Alternative techniques such as allogeneic skin grafts and tissue expansion may be used in rare circumstances.9,10
Care of the child with a myelomeningocele is life-long; it only begins with the surgical closure. Any deterioration in neurological function signals a progressive process such as shunt malfunction, hydromyelia, tethered cord, or symptomatic Chiari II malformation. Significant advancements have been made in the treatment of these children over the past two decades, particularly in the widespread use of multidisciplinary teams of specialists to manage their urological, orthopedic, and other needs. Among those who undergo early back closure, 92% will survive to 1 year.11 From prospective outcome cohort data, it is known that the survival rate until age 17 is 78%,12 but it drops to 46% by the fourth decade of life.13 Death is the result of problems associated with the Chiari II malformation, restrictive lung disease secondary to chest deformity, shunt malfunction, or urinary sepsis. A sensory level higher than T11 is associated with increased risk of mortality,13 likely due to increased risk of urosepsis.14 Approximately 75% of children with myelomeningocele are ambulatory, although most require braces and crutches. Approximately 75% of surviving infants will have normal intelligence (defined as IQ > 80), although only 60% of those requiring shunts for hydrocephalus will have normal intelligence.15 Normal intelligence drops to 70% in surviving adults.16
In a few centers, the fetus with spina bifida may be a candidate for in utero treatment, because this condition is routinely detected before 20 weeks of gestation. There is evidence that neurological deterioration occurs during gestation.17 Normal lower extremity movement can be seen on sonograms of affected fetuses before 17 to 20 weeks of gestation, but most late-gestation fetuses and newborns have some degree of deformity and paralysis. Such deterioration could be the result of exposure of neural tissue to amniotic fluid and meconium or direct trauma as the exposed neural placode impacts against the uterine wall. In theory, such deterioration could be reduced or eliminated by in utero closure of the lesion. Animal studies (in which a model for spina bifida is created by laminectomy and exposure of the fetal spinal cord to the amniotic fluid) have demonstrated improved leg function if the lesion is closed before birth.18 There is also evidence that the Chiari II malformation, which occurs in the vast majority of individuals with spina bifida, is acquired and could potentially be prevented by in utero closure.19
The first cases of in utero spina bifida repair were performed in 1994 using an endoscopic technique. This technique proved unsatisfactory and was quickly abandoned. In 1997, in utero repair of spina bifida was performed by hysterotomy at Vanderbilt University and at the Children’s Hospital of Philadelphia.20,21 Fetuses treated in utero are delivered by cesarean section because the forces of labor are likely to produce a uterine dehiscence. The early experience at both institutions suggested that relative to babies treated postnatally, those treated in utero had a decreased incidence of hindbrain herniation, and possibly a decreased need for shunting.22,23 The combined experience at the Children’s Hospital of Philadelphia and Vanderbilt, indicates that the incidence of hydrocephalus requiring shunting in patients treated in utero is less than in historical control subjects stratified by spinal level who received standard postnatal care.24,25 It is hypothesized that fetal closure of the spinal lesion reduces the need for shunting by eliminating the leakage of spinal fluid which puts back-pressure on the hindbrain. This allows reduction of the hindbrain hernia and relieves the obstruction of the outflow from the fourth ventricle.26
In utero spina bifida closure appears to be generally well tolerated by the expectant mothers. Approximately 5% of fetuses have died from complications associated with uncontrollable labor and premature birth. An analysis of leg function in children treated prenatally revealed no significant difference from a set of historical control subjects who were treated with conventional postnatal repair.27 However, many of the children evaluated in this series had lower limb paralysis at the time of the surgery, which may have diluted any possible benefit. In contrast, a series from the Children’s Hospital of Philadelphia suggested potentially improved leg function in patients with prenatally confirmed intact leg movement on ultrasound prior to fetal surgery.28 Problems with delayed development of dermoid inclusion cysts and tethered cord may adversely affect outcome in the long term.29 The preliminary experience suggests that children treated in utero have the same urodynamic abnormalities that are seen in conventionally treated children with spina bifida.30,31 The incidence of the Chiari II malformation, and the need for shunting may be decreased,23 but there are currently no long-term data.
Prior to the Management of Myelomeningocele Study (MOMS) trial,32 outcomes for spina bifida babies treated in utero were assessed relative to outcomes in conventionally treated, historical control subjects.8 Such comparisons are, however, prone to substantial biases because fetuses that undergo in utero closures represent a highly selected subset of cases. In addition, the medical management of spina bifida is continuously improving, making comparisons with historical control subjects particularly problematic.
A consortium of three institutions (Children’s Hospital of Philadelphia, Vanderbilt, and University of California San Francisco) performed an unblinded, randomized, controlled trial of in utero treatment of spina bifida ([MOMS] to obtain definitive answers regarding the benefits of fetal myelomeningocele closure.32). Pregnant women who receive a prenatal diagnosis of spina bifida between 16 and 25 weeks of gestation were randomized to either in utero repair at 19 to 25 weeks’ gestation or cesarean delivery after demonstration of lung maturity. The primary study end points were the need for a shunt procedure at 12 months, and fetal/infant death. Secondary end points included neurological function, cognitive outcome, and maternal morbidity. The intent to treat analysis demonstrated a significant risk reduction with regard to the primary endpoint, and the study was closed early due to efficacy of prenatal surgery. The prenatal surgery group benefited from decreased shunt requirement (40% versus 82%) and a higher proportion of normal hindbrain anatomy, and it was more likely to ambulate independently at 30 months compared to the postnatal group. There were no maternal deaths, and adverse neonatal outcomes were similar between groups; however, prenatal surgery was associated with more pregnancy complications, increased frequency of pre-term delivery, and a higher rate of respiratory distress syndrome in the neonate. To date, this is the only randomized study that demonstrates clear benefits of in utero treatment of spina bifida. These benefits were realized at experienced centers with strict inclusion criteria and must be carefully weighted against the higher rates of prematurity and maternal morbidity. Longer follow up is required to determine the longevity of these benefits as well as the effect on urinary function.
Occult Spina Bifida and the Tethered Cord Syndrome
The term occult spinal dysraphism actually encompasses several separate, possibly coexisting, entities. Most of these entities are localized to the lower spine segments and hidden by full-thickness skin. Embryologically, they arise from abnormal retrogressive differentiation of the caudal cell mass, a process by which the previously formed tail structures undergo a precise, ordered necrosis, leaving only the filum terminale, the coccygeal ligament, and the terminal ventricle of the conus as remnants by 11 weeks of gestation. Failure of regression presumably gives rise to the hypertrophied filum terminale; abnormal and incomplete regression result in lipomyelomeningocele. The embryology of diastematomyelia remains poorly understood,33 but it may involve persistence of the fetal neurenteric canal between the yolk sac and the amniotic cavity, allowing herniation of endodermal elements through a split notochord, and causing migrating mesenchymal elements to form the bony “spike.”
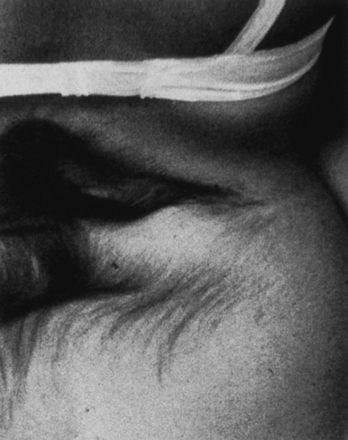
To recognize occult dysraphic states, one must appreciate the significance of the various syndromes that occur in association with the various entities (Table 5.1). The cutaneous syndrome refers to any midline skin anomaly overlying the lower spine. This anomaly often signals a dysraphic state, and its recognition is especially important in the infant, in whom urological or orthopedic complaints are not yet manifest. Dimples may be significant if they are at the level of the upper sacral or lumbar spine above the gluteal fold, but the common coccygeal pit overlying the lowest point of the coccyx in or below the gluteal fold has no particular significance.34,35 The cutaneous abnormality may include the striking “faun’s tail” of hair (Fig. 5.7), dermal sinus tract, hemangioma (Fig. 5.8), or skin-covered fatty mass (Fig. 5.9). The orthopedic syndrome is apparent at birth or develops progressively in childhood. Common components include high arched feet, claw toes, unequal leg length, and scoliosis. The urological syndrome should be considered in any infant or small child who has an abnormal voiding pattern, a child with a new onset of incontinence after toilet training, or with urinary tract infection in a child of any age. The neurological syndrome presents as leg muscle atrophy or weakness, numbness of the feet, or radicular lower extremity pain and can occur at any age. In summary, patients may present with any of the abovementioned syndromes, but in general, infants primarily present with skin manifestations, older children present with urological, neurological, or orthopedic syndromes, and adults often complain of pain (see Table 5.1).36
TABLE 5.1 Presenting Symptoms and Signs of Occult Spinal Dysraphism
| Symptoms/Signs | Frequency |
|---|---|
| Foot deformity | 39% |
| Scoliosis | 14% |
| Gait abnormality | 16% |
| Leg weakness | 48% |
| Sensory abnormality | 32% |
| Urinary incontinence | 36% |
| Recurrent urinary tract infections | 20% |
| Fecal incontinence | 32% |
| Cutaneous abnormality | 48% |
Adapted from Pang D: Sacral agenesis and caudal spinal cord malformations. Neurosurgery 1993;32:755-758.
< div class='tao-gold-member'>
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


