CHAPTER 38 SPINE AND SPINAL CORD: DEVELOPMENTAL DISORDERS
Developmental disorders of the spine and spinal cord occur because of defects in the development and maturation of the nervous system. A basic understanding of embryology is therefore important in appreciating the nature and scope of these disorders. There are various ways of approaching and thinking about the developmental disorders:
As knowledge and understanding of these disorders continue to expand, the most reasonable approach is to integrate advances in genetic and etiological models within the traditional phenotypic clinicoanatomical framework. Developmental disorders can be classified anatomically, depending on whether they affect primarily the spinal cord and neural tissues or the spine and supporting structures (Table 38-1).
TABLE 38-1 Developmental Disorders That Can Affect Vertebrospinal Structures
| Disorders Affecting the Spine | Disorders Affecting the Spinal Cord and Neural Tissues | Vascular Malformations |
|---|---|---|
| A variety of vascular malformations can affect the spine and spinal cord | ||
| Scoliosis | Developmental cysts |
NORMAL DEVELOPMENT OF THE NERVOUS SYSTEM
The human embryonic period spans the first 8 postfertilization weeks and is classified on the basis of a morphological system. There are 23 distinct Carnegie stages, each stage covering a period of 2 to 3 days. This staging system facilitates an understanding of the timing and sequence of embryonic development. The fetal period spans the 9th week after fertilization to birth. Fetal age is thought of mainly as measurements, inasmuch as a similar morphological staging system is not available. The great majority of congenital malformations begin during the embryonic period.1–3
The neural tube is the embryonic structure that develops into the brain and spinal cord. Primary neurulation refers to the development of the neural tube from the neural plate. Complete development and closure of the neural tube occurs from days 17 to 30 after conception and spans Carnegie stages 8 to 12 in the embryonic period (Table 38-2). The caudal eminence is the continuation of the primitive streak and develops into the terminal portions of the notochord, somites, vertebrae, and hindgut. The neural cord arises from the caudal eminence and forms the caudal portion of the spinal cord. This process is secondary neurulation. The level of the junction between primary and secondary neurulation is at the level of the future second sacral vertebra. There is elongation of the previous neural tube together with formation of the lower sacral and coccygeal segments. This part of the development of the nervous system commences in stage 12 at 4 weeks and ends approximately at stage 20.2–6
TABLE 38-2 Important Stages in Primary Neurulation and the Formation and Closure of the Neural Tube
| Development Stage No. | Days in Embryonic Life | Nervous System Development |
|---|---|---|
| 8 | 17-19 | Development of the neural plate |
| 9 | 19-21 | Development of the neural folds |
| 10 | 22-23 | First steps in the fusion of the neural folds: There are two initial sites of fusion: fusion proceeds bidirectionally from the lower medullary rhombencephalic site and in a caudal direction from the higher prosencephalic site. The fusions terminate in two neuropores: the rostral and caudal neuropores. |
| 11 | 23-26 | Closure of the rostral neuropore |
| 12 | 26-30 | Closure of the caudal neuropore |
Based on a table from Lemire RJ: Neural tube defects. JAMA 1988; 259:558-562 Copyright © 1988 American Medical Association. All rights reserved; and on information from O’Rahilly R, Muller F: The two sites of fusion of the neural folds and the two neuropores in the human embryo. Teratology 2002; 65:162-170.
The vertebral bodies of the spine are generated from the mesenchymal cells. The process of gastrulation occurs at day 14 in embryonic life to generate the mesenchymal cells that will in turn develop into the head, cardiac, and the paraxial and lateral mesoderm. At 20 to 30 days in embryonic life, the paraxial mesoderm, in a process of somatogenesis, subdivides into spherical somite segments on each side of the spinal cord in a rostral-to-caudal direction. The somites first appear in stage 9; differentiation commences at stage 10. The somites mature and further subdivide into the sclerotome to form the vertebral bodies, the myotome to form the musculature, and the dermatome to form the dermis. The sclerotome undergoes a resegmentation process in which the caudal end of one somite links with the rostral end of the next somite to form a vertebral body.7–9
DEVELOPMENTAL DISORDERS AFFECTING THE SPINE
Klippel-Feil Anomaly
Epidemiology
The Klippel-Feil anomaly occurs in 1 per 40,000 to 1 per 42,000 births, with a female-to-male ratio of 3:2.8
Clinical Features
Individuals with Klippel-Feil anomaly may be asymptomatic. Clinical manifestations can range from pain, through orthopedic or neurological manifestations, to a more widespread malformation disorder. The initial description was of a classical triad of shortening of the neck, limited range of neck movement, and a low posterior hairline. Affected individuals may complain of cervical pain and present with cosmetic deformities. The Klippel-Feil anomaly may be associated with spinal instability. This occurs if there are unstable fusion patterns or if stenosis and arthritis develop at the interspaces between the fused joints. The associated axial and spine anomalies include cervical or fused ribs, cleft vertebrae or hemivertebrae, and kyphoscoliosis.8
The Klippel-Feil anomaly may be associated with other spinal developmental disorders, including syrinx, tethered cord, and split cord malformations (SCMs). Alternatively, the Klippel-Feil anomaly could be part of a widespread multisystem developmental disorder as well, in which other features include hearing deficits, congenital heart disease, and genitourinary manifestations.8
Etiology and Pathophysiology
The Klippel-Feil anomaly results from failure of normal segmentation of the vertebral column in early embryonic life. The exact cause and mechanism behind this syndrome are unclear; the Klippel-Feil anomaly may result from disruptions in isolated genes that govern segmentation, or it may result from complex gene-gene or gene-environment interactions.8
Management
Diagnostic investigations center on imaging studies. Good-quality radiographs and computed tomographic (CT) scans of the cervical spine are necessary to show the extent of cervical fusion and to evaluate potential instability (Fig. 38-1). Magnetic resonance imaging (MRI) studies, including flexion and extension views, are useful for evaluating instability or stenosis in the cervical spine and for assessing neurocompression of the spinal cord. MRI should be performed on the whole neuraxis in order to look for the associated developmental anomalies. Further evaluation is targeted at the associated conditions, including audiology assessments and cardiac and renal investigations.8
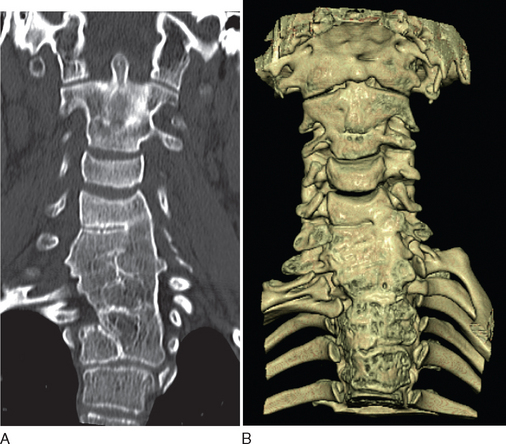
Management depends on the symptoms and the presence of other malformations. Treatment modalities may include simple measures of modifying activities and the use of traction and bracing to alleviate deformities. Surgical intervention to stabilize the spine is indicated if there is neurological impairment or symptomatic cervical instability. Surgical options include occipitocervical arthrodesis and combined atlantoaxial and subaxial arthrodesis.8
Basilar Impression
Definition
Basilar impression (or basilar invagination) occurs when there is an abnormal upward displacement of the basilar and condylar portions of the occipital bone. This leads to invagination of the foramen magnum into the posterior cranial fossa with associated translocation of the upper cervical vertebrae into this depression. We believe the terms basilar impression and basilar invagination are interchangeable, but some authors differentiate basilar impression from basilar invagination on the basis of differences in anatomical definitions and causation (e.g., acquired or primary).10,11 Some texts include platybasia in the spectrum of basilar impression. Platybasia is an abnormal flattening of the base of the skull with an abnormally obtuse angle between the plane of the clivus and the plane of the anterior fossa. This may occur together with basilar impression but is of no real clinical significance.
For instance, if Chamberlain’s line is the defining parameter, basilar impression is present when more than one third of the odontoid process lies above this line.10,12
Epidemiology
Basilar impression is the most common of the craniocervical malformations; clinical manifestations usually occur from the second decade onward.11 The true incidence is unclear.
Clinical Features
Basilar impression may be asymptomatic or may manifest with deformities, including a shortened neck. Movement or exercise-induced occipital headache is a common feature. Oculomotor features include horizontal and upward-beat or downward-beat nystagmus. Other cranial nerve symptoms include facial nerve spasm, trigeminal sensory symptoms, and trigeminal neuralgia. Pontomedullary compression can lead to pyramidal tract signs, with the lower limbs typically more affected than the upper limbs. Sleep apnea and respiratory depression have been described.11,12
Primary basilar impression is often associated with other developmental anomalies, including occipitalization of the atlas, the Klippel-Feil anomaly, the Arnold-Chiari malformation, and syringomyelia or syringobulbia.12
Etiology and Pathophysiology
Management
The treatment goal is to relieve current compression, and to prevent future compression, of the neuraxis. The treatment modalities for symptomatic basilar impression include cervical traction and surgical intervention. Cervical traction has been reported to cause vertebral artery dissection, as well as potentially causing infection, cranial fractures, and hemorrhage. Anterior compression is managed with an anterior surgical approach; posterior or circumferential compression is managed with a posterior approach. The role of prophylactic surgery or prolonged immobilization with asymptomatic lesions is unclear.10,12
Occipitalization of the Atlas
Clinical Features
This condition may be asymptomatic. However, atlantoaxial dislocation can occur in up to 60% of affected patients. In this condition, there is excessive compensatory stress on the atlantoaxial joint as a result of the decreased movement of the atlantooccipital joint. This situation increases the possibility of developing traumatic dislocation or cervical degenerative joint disease. Occipitalization of the atlas is associated with other developmental disorders as well, including basilar impression, Klippel-Feil anomaly, and Chiari malformations.13
Atlantoaxial Dislocation
Definition
Epidemiology
The various degrees of C1 malformations have a quoted prevalence rate of 4%, which is based on autopsy studies.14
Clinical Features
Abnormalities in this region may be asymptomatic and found incidentally, or they may manifest with pain. The vertebral anomalies may lead to atlantoaxial instability and dislocation. This may result in cervicomedullary compression, which may manifest either spontaneously or after trauma. Affected individuals are especially prone to cord compression during extension injuries: for instance, with intubation or after neck injuries. These anomalies should be considered when a child presents with a neurological deficit or neck pain after minor trauma.14,15
Congenital atlantoaxial instability may be seen in association with Down’s syndrome. Other congenital craniovertebral junction and spinal disorders may occur in tandem as well, including Chiari type I and SCMs. It is important to be aware that atlantoaxial instability may not be congenital in nature but may be caused by an underlying condition, such as rheumatoid arthritis.14,15
Etiology and Pathophysiology
The craniovertebral junction malformations arise from defects during the ossification process. The malformations lead to instability in the atlantoaxial joint with subsequent dislocation. Subluxation usually occurs in the horizontal plane, with C1 moving anteriorly to C2. In other settings, progressive cervical myelopathy rarely occurs above C2; the usual scenario of a myelopathy is in the context of acquired degenerative change and usually occurs below C3. The risk of neurocompression depends on the underlying malformation and the joint stability. The actual degree of neurocompression depends on the extent of the subluxation and the width of the spinal canal. The normal sagittal diameter of the spinal canal is 16 to 25 mm at the level of the atlas; the spinal cord is at risk when the canal diameter is less than 14 mm.14
Management
The use of high-quality imaging with plain radiographs, CT scans (including fine cuts), and MRI is necessary in the evaluation of atlantoaxial dislocation and craniocervical anomalies. Flexion and extension views are especially useful for assessing joint stability. MRI is crucial in the assessment of the neural structures, especially with regard to cord compression. Surgery is indicated in order to decrease the possibility that significant neurological deficits may develop and should be considered when there is neurocompression or when the individual is symptomatic. Surgery for atlantoaxial and craniocervical instability is a challenging field, especially because surgeons have to work with abnormal anatomy while taking into account the ongoing growth potential for the child. Depending on the underlying malformation, surgical management consists of decompression procedures and joint fusion and stabilization.14–17
Scoliosis
Epidemiology
Idiopathic scoliosis affects 2% to 4% of children aged 10 to 16 years. The incidence is equal in boys and girls for curves of less than 20 degrees. Girls, however, significantly outnumber boys, at 5:1, for curves of more than 20 degrees. Secondary neurogenic scoliosis is more common in the younger population. For instance, of patients with the Chiari malformation and syringomyelia, 82% younger than 20 years have scoliosis, as opposed to only 16% of those older than 20 years.18,19
Clinical Features
Individuals with scoliosis may present with back pain or, more obviously, with the spinal curvature deformity. In evaluating scoliosis, it is imperative to be aware that the spinal deformity may be the presenting feature of an underlying spinal cord or brainstem anomaly in 4% to 58% of affected patients. Individuals with scoliosis may present with the neurological or orthopedic features of the underlying developmental disorder.18,19
Etiology and Pathophysiology
Secondary scoliosis is thought to arise from a generalized paresis of truncal musculature or congenital changes in the vertebrae. In addition, scoliosis in the Chiari-syringomyelia complex may be caused by the abnormal intramedullary pressures within the spinal cord that interfere with the postural tonic reflexes.18,19
Management
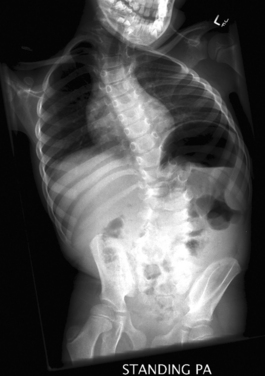
It is important to treat or rule out an underlying spinal cord or brainstem disorder in a young patient with scoliosis. Evaluation includes the use of radiographs (Fig. 38-2) to chart the pattern of scoliosis and MRI to look for associated anomalies in the neural elements. Posterior fossa decompression for the Chiari-syringomyelia malformation can lead to an improvement in the scoliosis, especially if done early in the patient’s life. Initial treatment may involve use of a spinal orthotic brace as well. Spinal fusion surgery may ultimately be required for surgical correction of the scoliosis.18,19
DEVELOPMENTAL DISORDERS AFFECTING THE SPINAL CORD AND NEURAL TISSUES
Chiari Malformation
Definition
In 1891, Hans Chiari first described the disorder that now bears his name as cerebellar tonsillar herniation below the plane of the foramen magnum into the spinal canal. Further contributions and observations were made by Julius Arnold and John Cleland. Chiari malformations are also known as cerebellar ectopy and are classified into four types:
Epidemiology
Chiari type I malformations have a prevalence of 0.6% to 0.9% in studies based on MRI. Symptomatic Chiari type I malformations are slightly more common in girls and women, with a female-to-male ratio of 1.3:1 to 1.7:1.22,23
Clinical Features
As with the other developmental disorders, the Chiari malformation may be asymptomatic or, alternatively, may be symptomatic with a wide spectrum of clinical manifestations. A very common manifestation is headaches with or without cervical pain. The headache is typically a protracted occipital-suboccipital headache and is exacerbated by the Valsalva maneuver, postural changes, and coughing or straining. Another common complaint is of weakness and altered sensation, including paresthesia and dysesthesias. A myelopathy may manifest with the sensorimotor or sphincter disturbances. Individuals with Chiari malformations may present with ataxia and other cerebellar signs. Downward-beating nystagmus and other oculomotor disturbances may occur. Other features of brainstem dysfunction may result, including cranial neuropathies, neuro-otological symptoms, sleep apnea, and dysphagia.21–24
Syringomyelia occurs in 32% to 74% of individuals with Chiari type I malformations. A cervical syrinx may manifest with upper limb neurological deficits, whereas a thoracic syrinx may lead to scoliosis. In addition, osseous anomalies of the base of skull, including basilar impression, may be seen with Chiari type I malformations. Chiari type II malformations are often associated with an NTD, other brainstem deformities, and hydrocephalus. Features of raised intracranial pressure are commonly the clinical manifestations of the associated hydrocephalus. The Chiari type I malformation is usually asymptomatic in childhood and tends to manifest in the second to third decade of adulthood. The Chiari type II malformation, however, is usually evident in childhood.21,23,24
Etiology and Pathophysiology
The Chiari malformation probably arises from underdevelopment of the occipital enchondrium, which leads to an undeveloped occipital bone with a small and shallow posterior fossa. This in turn leads to overcrowding of the posterior fossa and a downward herniation of the brain. The occipital enchondrium originates from the occipital somite, which in turn is derived from paraxial mesoderm. If the occipital enchondrium is more severely affected, basilar impression may result as well. The degree of tonsillar herniation is thought to be correlated to some extent with the severity of symptoms.21,24
Management
The main investigation for a suspected Chiari malformation is MRI of the neuraxis. Sagittal views are particularly important in confirming the diagnosis and demonstrating the degree of cerebellar and brainstem herniation and compression (Fig. 38-3). MRI is also useful for identifying the associated hydrocephalus or syrinx and identifying other developmental disorders, including NTDs, although these are usually obvious clinically.
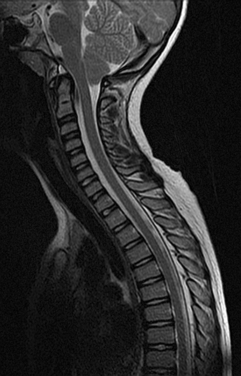
If there are neurological symptoms or signs, surgical intervention is usually indicated, to stabilize or ameliorate symptoms. The goal of surgery is to relieve brainstem and cerebellar compression. Ventral compression can be surgically treated with a transoral clivus odontoid resection. If there is no ventral compression, posterior fossa decompression is usually undertaken. A ventriculoperitoneal shunt may be inserted for associated hydrocephalus. The syrinx usually improves after recovery of cerebrospinal fluid (CSF) flow dynamics. If indicated, especially if the syrinx persists despite decompression, a shunt procedure for the syrinx can be undertaken as well.18,19






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


