Chapter 11 Stiffness syndromes
Muscle stiffness may be the presenting symptom in many disorders of the motor nervous system and muscles (Table 11.1) (Rowland, 1985; Thompson, 1993, 1994; Brown and Marsden, 1999). Spasticity is the most classic, and the others must be distinguished from it. In this chapter, therefore, spasticity will be considered first. The stiff-person syndrome and its variants is perhaps the major entity confronting the movement disorder specialist, and it will be considered next. Stiffness can arise not only from dysfunction of the central nervous system but also from disorders where there is continuous muscle activity where the pathology lies in the muscle, nerve, or anterior horn cell. These conditions will be considered last.
Table 11.1 Causes of muscle stiffness, cramps, spasms, rigidity, or contracture
| Cerebral – brainstem |
| Spinal cord |
| Peripheral nerve |
| Muscle |
| Unknown origin |
| Contracture |
Stiffness is assessed by the amount of force needed to get a movement. Tone, the more general clinical term, can be defined as the resistance to passive stretch of a joint. Normal tone is very low, and it is difficult to appreciate a decrease in tone, but some authorities say that they can detect a hypotonia in cerebellar dysfunction. Increased tone, hypertonia, is characteristic of several different states and can come from three theoretical mechanisms: (1) altered mechanical properties of the muscle or joint; (2) background co-contraction of muscles acting on the joint; and (3) increase in reflex response to the stretch opposing the movement (Hallett, 1999, 2000). A clear example of altered mechanical properties is contracture. Increased background contraction is often seen with difficulty in relaxation, such as commonly characterizes Parkinson’s disease. There are many different reflexes that can occur in response to stretch and these can help differentiate different states of hypertonia. In differentiating spasticity and rigidity, in simple terms, spasticity shows exaggerated short latency reflexes and rigidity shows exaggerated long latency reflexes.
Spasticity
Spasticity is a form of hypertonia with a number of characteristic features (Benecke et al., 2002; Sheean, 2002; Sanger et al., 2003). The increased resistance to stretch is velocity sensitive; there is more resistance the faster the joint is moved. There may be a clasped-knife phenomenon where the resistance increases and then suddenly gives way. There are also several other “positive” features such as increased tendon jerks, clonus, increased flexor reflexes, spontaneous flexor spasms, and abnormal postures (spastic dystonia). Importantly, there are also “negative” features including weakness (in a “pyramidal distribution”), fatigue, loss of coordination, and a decrease of some cutaneous reflexes. It can be noted that the increased stiffness can be valuable to a patient with significant weakness, as it might, for example, allow the patient to stand.
Loosely, neurologists usually say that spasticity arises from a lesion in the pyramidal tract or that it is from damage to the “upper motor neuron.” The first seems false and the second is vague. Supraspinal control of movement is complicated and consists of many tracts. Briefly, those fibers that go through the pyramid arise from the cortex and go to the spinal cord and can be called also the corticospinal tract (Wiesendanger, 1984; Davidoff, 1990). Approximately 30% of those fibers arise from the primary motor cortex; there are also significant contributions from premotor cortex and sensory cortex. The fibers largely cross in the pyramid, but some remain uncrossed. Some terminate as monosynaptic projections onto alpha-motoneurons, and others terminate on interneurons including those in the dorsal horn. Other cortical neurons project to basal ganglia, cerebellum, and brainstem; these structures can also originate spinal projections. Particularly important is the reticular formation that originates several tracts with different functions (Nathan et al., 1996). The dorsal reticulospinal tract may have particular relevance for spasticity and is normally inhibitory onto the spinal cord (Habaguchi et al., 2002; Takakusaki et al., 2001, 2003). Thinking about the cortical innervation of the reticular formation, it is possible to speak of a cortico-reticulo-spinal tract. Lesions of the primary motor area alone and lesions of the pyramid alone do not cause spasticity (Sherman et al., 2000). It appears that premotor damage is necessary and likely involvement of cortico-reticulo-spinal pathways. Dysfunction of the dorsal reticulospinal tract will disinhibit the spinal cord and may give rise to the hyperexcitability characteristic of spasticity. The term “corticofugal syndrome” has been suggested to indicate that “spasticity” has important negative as well as positive features and that the lesions involve descending tracts other than the corticospinal tracts, but it is not commonly accepted (Thilmann, 1993).
Clinical features of spasticity that help with the diagnosis, in addition to the velocity-dependent increased tone, include brisk tendon reflexes, the Babinski sign, Hoffman reflex (indicating brisk finger flexor reflexes), and loss of cutaneous abdominal reflexes. The negative features will often be seen as well, with weakness in the lower extremities of flexors more than extensors and in the upper extremities of extensors more than flexors. In the clinical neurophysiology laboratory, there will be increased H reflexes, identified with an increase of the maximum amplitude H reflex compared to the M wave (muscle response to direct supramaximal stimulation of the nerve), called the H/M ratio (Hallett, 1999, 2000; Benecke et al., 2002). There is also a diminished decrease of the H reflex with vibration of the body part. Characteristics of the tonic stretch reflex can also be assessed for threshold and gain to stretches of varying velocity. In spasticity, there is some controversy, but both lowered velocity threshold and an increased gain have been found (Powers et al., 1988; Katz and Rymer, 1989; Thilmann et al., 1991; Ibrahim et al., 1993; Musampa et al., 2007). It is important to recognize that there are both reflex and nonreflex contributions to spastic hypertonia (Chung et al., 2008).
There are many methods to treat spasticity, but this must be done carefully as correction of the positive features may not be all that helpful, and, as noted before, may even be detrimental. For many patients, the much more important aspects of their corticofugal syndrome are the negative features such as the weakness and these cannot be dealt with easily. Increased tone can be improved with a variety of oral agents including benzodiazepines, baclofen, and tizanidine (Krach, 2001; Abbruzzese, 2002; Ronan and Gold, 2007; Kamen et al., 2008). Baclofen can be given intrathecally by pump, and this can be much more efficacious, likely because of the ability to increase the dose at the target tissue without side effects (Ivanhoe et al., 2001; Albright et al., 2003; Dykstra et al., 2007; Rietman and Geertzen, 2007; Saval and Chiodo, 2010). Tolperisone has been evaluated in patients after stroke (Stamenova et al., 2005) and might be another consideration (Quasthoff et al., 2008).
Direct blockade of muscle contraction with agents such as phenol has been used for some time and the introduction of botulinum toxin for this purpose has been welcomed with enthusiasm (Boyd and Hays, 2001; Moore, 2002; Barnes, 2003; Mancini et al., 2005; Mohammadi et al., 2009). An evidence-based review gave botulinum toxin its highest recommendation for treatment in both adults and children (Simpson et al., 2008). For post-stroke spasticity, there is very good evidence for reduction of spasticity. Evidence is developing for an increase in functional ability (Elia et al., 2009; McCrory et al., 2009; Foley et al., 2010; Fridman et al., 2010; Sun et al., 2010), but often this is rather limited (Kaji et al., 2010). Children with cerebral palsy can be much improved (Baird and Vargus-Adams, 2009; Kanellopoulos et al., 2009; Lukban et al., 2009; Coutinho Dos Santos et al., 2011), but are a vulnerable population and need to be treated with care (Albavera-Hernandez et al., 2009). Several evidence-based reviews document the utility of botulinum toxin for this indication (Delgado et al., 2010; Hoare et al., 2010). It is interesting to note that the effect of botulinum toxin is mediated not only on the alpha motoneuron neuromuscular junction but also via the gamma motoneuron effect on the muscle spindle (Trompetto et al., 2008). Surgical methods such as rhizotomy can also be used in some cases for symptomatic relief of severe spasticity (Lazorthes et al., 2002).
Stiff-person (stiff-man) syndrome
The stiff-person syndrome (originally called the stiff-man syndrome) (Table 11.2) consists of progressive fluctuating muscular rigidity (Moersch and Woltman, 1956; Blum and Jankovic, 1991; Thompson, 1993, 1994; Stayer and Meinck, 1998; Brown and Marsden, 1999; Levy et al., 1999; Thompson, 2001; Meinck and Thompson, 2002). Typically, the rigidity affects axial muscles of the back, abdomen, hips, and shoulders, causing excessive lordosis with prominent contraction of paraspinal muscles, a “board-like” abdomen, and stiffness of the legs on walking (Fig. 11.1) (Video 11.1). Superimposed upon this continuous stiffness are spasms provoked by excitement, anxiety, voluntary movement, sudden noise, or peripheral stimuli. These spasms can be intensely painful and forceful such as to fracture bones or dislocate joints. Voluntary movement can provoke similar spasms that sometimes may cause falls “like a wooden man.” 
Table 11.2 Criteria for the diagnosis of the stiff-person syndrome
| Clinical |
| Neurophysiologic |
| Normal motor unit morphology |
| Other observations that may be helpful but are of uncertain diagnostic specificity |
Association with autoimmune endocrine disease (e.g., diabetes, pernicious anemia, vitiligo, hypothyroidism) |
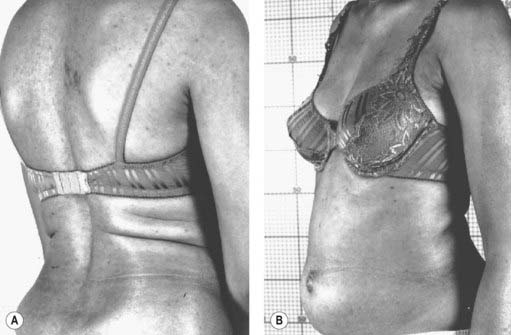
Figure 11.1 Patient with stiff-person syndrome. Note the marked lumbar lordosis (A) and the prominent abdomen (B).
Photos courtesy of Dr M. Dalakas.
The syndrome usually begins in the fourth and fifth decades and affects men and women equally. The onset of the illness usually is gradual with increasing painful tightness, stiffness, clumsiness of the trunk and legs, and limitation of range of motion (Fig. 11.2). On examination there is continuous muscular contraction of the paraspinal and abdominal muscles, but there are no other neurologic signs, other than brisk reflexes. The illness is slowly progressive with stiffness spreading from the trunk to hip and then shoulder muscles, but the face and distal limbs usually are spared. Sphincter function is normal. While the onset of the disorder seems typically spontaneous, one case has been reported where the onset appears to have been triggered by West Nile fever (Hassin-Baer et al., 2004). There has also been a report of a father and daughter, each with anti-GAD-positive stiff-person syndrome (Burns et al., 2003).

Figure 11.2 Patient with stiff-person syndrome. Note the limitation in bending forward.
Photo courtesy of Dr M. Dalakas.
Electromyography shows continuous normal motor unit activity, despite attempted relaxation, with no signs of denervation and normal peripheral motor and sensory nerve conduction velocity. Other physiologic abnormalities include exaggerated, non-habituating exteroceptive or cutaneomuscular reflexes, brainstem myoclonus, and an exaggerated startle reflex (“jerking stiff-man”) (Leigh et al., 1980; Meinck et al., 1983; Matsumoto et al., 1994; Meinck et al., 1995; Stayer and Meinck, 1998; Brown and Marsden, 1999).
Insulin-dependent diabetes mellitus occurs in up to two-thirds of patients. Diabetic ketoacidosis is the commonest cause of death in such patients. Other autoimmune endocrine diseases include thyroid disease, pernicious anemia, and vitiligo (Solimena et al., 1990). Epilepsy occurs in 10% of cases, although this is debatable.
A central, perhaps spinal cord origin for the spasms, rigidity, and continuous motor unit activity is suggested by their disappearance after peripheral nerve block, sleep, and general anesthesia. Other neurophysiologic tests suggest that spinal motoneuron excitability is normal and that the condition is due to defective input of inhibitory pathways onto motoneurons. To test inhibitory spinal circuits in patients, Floeter et al. (1998) used H-reflexes to test reciprocal inhibition in the forearm and thigh, vibration-induced inhibition of flexor carpi radialis and soleus H-reflexes, recurrent inhibition, and nonreciprocal (1b) inhibition of soleus H-reflexes. Vibration-induced inhibition of H-reflexes was diminished in eight of nine patients tested, but the presynaptic period of reciprocal inhibition was normal in most patients. Both circuits are presumed to involve presynaptic inhibition and GABAergic interneurons. Presumed glycinergic circuits, including the first period of reciprocal inhibition and nonreciprocal (1b) inhibition, showed occasional abnormalities. Recurrent inhibition was normal in all five patients tested. It appears that some, but not all, populations of GABAergic neurons are affected. The involvement of presumptive glycinergic circuits in some patients could point to impairment of non-GABAergic neurons, unrecognized involvement of GABAergic neurons in these inhibitory circuits, or, more likely, alterations of supraspinal systems that exert descending control over spinal circuits. Studies of cortical excitability with transcranial magnetic stimulation show decreased intracortical inhibition likely due to loss of GABA function at this level (Sandbrink et al., 2000). Hyperexcitability of the brainstem has been demonstrated by increased R2 recovery in the blink reflex recovery curve (Molloy et al., 2002). Direct measurement of GABA with magnetic resonance spectroscopy shows a deficiency in the sensorimotor cortex (Levy et al., 2005). There is also a cortical loss of flumazenil binding, a measure of postsynaptic benzodiazepine receptors associated with GABA receptors (Perani et al., 2007; Galldiks et al., 2008).
The significance of the association of insulin-dependent diabetes mellitus with the stiff-person syndrome has been emphasized by the discovery of antibodies directed against glutamic acid decarboxylase (GAD), the enzyme responsible for the synthesis of GABA, in both blood and cerebrospinal fluid in 60% or more of patients (Solimena et al., 1990; Walikonis and Lennon, 1998). The great majority of these patients also have antibodies directed against pancreatic islet cells, as well as gastric parietal cells and the thyroid. The anti-GAD antibodies are the same as those found in insulin-dependent diabetes mellitus, but it appears that the antibodies do have subtle differences (Lohmann et al., 2000). Currently the best test is the radioimmunoassay for GAD65 (Walikonis and Lennon, 1998). The luciferase immunoprecipitation technology (LIPS) seems also to be very good for this purpose (Burbelo et al., 2008). Present evidence raises the possibility that the anti-GAD antibodies may destroy GABAergic inhibitory mechanisms in the spinal cord, but, alternatively, they may be secondary to some other process leading to an appropriate immunologic response (Raju et al., 2005; Raju and Hampe, 2008). Anti-GAD antibodies have been demonstrated to block GABAergic neurotransmission in rat cerebellar slices (Ishida et al., 1999; Mitoma et al., 2000). One study reported that motor cortex excitability correlated with antibody levels suggesting an etiologic role (Koerner et al., 2004). In a study of 18 patients with stiff-person syndrome and serum antibodies, all had high titers as well in the cerebrospinal fluid (CSF) and 11 of 13 patients had an increased anti-GAD(65)-specific IgG index (Dalakas et al., 2001b). In the same study, the mean level of GABA in the CSF was found to be lower in patients than in controls. On the other hand, the levels of antibodies do not correlate with the severity of the disorder (Rakocevic et al., 2004).
The role of anti-GAD antibodies continues to be somewhat confusing, in part because they are not specific to stiff-person syndrome. In one study of 61 neurologic patients with high antibody titers, 22 (36%) had stiff-person syndrome and 17 (28%) had cerebellar ataxia (Saiz et al., 2008). Intrathecal synthesis of antibody gives more assurance that the antibody might be relevant (Jarius et al., 2010).
Another antibody found in a large number of patients is that directed against GABA(A) receptor-associated protein (GABARAP) (Raju et al., 2006; Dalakas, 2008, 2009). This has been identified in up to 65% of patients. Such antibodies would inhibit GABA(A) receptor expression on GABAergic neurons and certainly would be pathophysiologically relevant.
Stiff-person syndrome can also be seen in association with anti-amphiphysin I antibodies in patients with breast cancer (Saiz et al., 1999; Wessig et al., 2003). Anti-amphiphysin antibodies can block GABAergic neurotransmission (Geis et al., 2010). A report of 11 patients with anti-amphiphysin antibodies found that all were women, and 10 had breast cancer (Murinson and Guarnaccia, 2008). Average age was 60, and cervical involvement was particularly prominent. Marked improvement was seen in three of five patients with tumor excision and chemotherapy. Other neurologic disorders such as sensory neuropathy, cerebellar ataxia, and opsoclonus may be present as well, and the syndrome can occur with other tumor types (Antoine et al., 1999). Another antibody in some cases is directed against 17-beta-hydroxysteroid dehydrogenase type 4 (Dinkel et al., 2002).
Oligoclonal bands have been reported in the CSF in a number of cases (Meinck and Ricker, 1987; Williams et al., 1988; Meinck et al., 1994), and white matter lesions have been seen on brain MRI. However, so far no consistent pathology has been demonstrated in the few cases that have come to autopsy.
The treatment of this condition relies upon a combination of benzodiazepines and baclofen in high dosage. These drugs may decrease the superimposed severe spasms, but are not entirely effective in controlling the background sustained continuous muscle hyperactivity. Sodium valproate and tizanidine have also been reported to be beneficial (Meinck and Conrad, 1986; Stayer and Meinck, 1998). Intrathecal baclofen has been used (Silbert et al., 1995; Stayer et al., 1997; Stayer and Meinck, 1998). Patients have been reported to respond to steroid therapy (Blum and Jankovic, 1991), intravenous human immunoglobulin (IVIg) infusions (Khanlou and Eiger, 1999; Souza-Lima et al., 2000), and plasmapheresis (Vicari et al., 1989; Blum and Jankovic, 1991; Brashear and Phillips, 1991; Hayashi et al., 1999), but others have gained no benefit from plasmapheresis (Harding et al., 1989). A double-blind, placebo-controlled study documented the value of IVIg (Dalakas et al., 2001a). Another study has shown improvement in quality of life with IVIg (Gerschlager and Brown, 2002). Rituximab has been reported to be useful in a few patients (Bacorro and Tehrani, 2010; Dupond et al., 2010; Katoh et al., 2010).
Progressive encephalomyelitis with rigidity, sometimes known as spinal interneuronitis, may present with similar clinical features to the stiff-person syndrome. However, such patients go on to develop a relentless and progressive course, with the emergence of cranial nerve dysfunction producing bulbar symptoms and disorders of eye movement, along with cognitive impairment and long tract signs (Whiteley et al., 1976; Howell et al., 1979; Brown and Marsden, 1999; Gouider-Khouja et al., 2002). The condition may be isolated, or may occur in the setting of neoplasia associated with the pathologic changes of paraneoplastic encephalomyelitis (Roobol et al., 1987; Bateman et al., 1990).
The condition may start at any age in adults (Kraemer and Berlit, 2008). Initial symptoms may be those of pain, dysesthesia or sensory loss in the limbs, or weakness, stiffness, clumsiness, and rigidity. Extensor trunk spasm and/or brainstem myoclonus may be striking (Video 11.2). The tendon reflexes often are absent and the plantar responses are extensor. Nystagmus, opsoclonus, ophthalmoplegia, deafness, dysarthria, and dysphagia can occur. The illness usually leads to death within about 3 years. As in the stiff-person syndrome, there is continuous motor unit activity with particular involvement of trunk muscles, which disappears after a peripheral nerve or spinal nerve root block, or general anesthesia. EMG exploration may reveal evidence of denervation of muscles. A few have reticular reflex myoclonus. Progressive myoclonus with rigidity and myoclonus has been called PERM. Some of these patients may also exhibit anti-GAD (Burn et al., 1991), or antineuronal (anti-Ri) antibodies (Casado et al., 1994). The CSF may contain a lymphocytic pleocytosis, elevated protein and immunoglobulin levels, and oligoclonal IgG bands. MRI may show brainstem atrophy and altered signal in the brainstem and spinal cord. 
Pathologic examination has shown widespread encephalomyelitis with perivascular lymphocytic cuffing and infiltration, associated with neuronal loss throughout the brainstem and spinal cord, mainly involving interneurons. The relation of progressive encephalomyelitis with rigidity to the classic stiff-person syndrome, particularly in those with anti-GAD antibodies, remains to be established (Brown and Marsden, 1999). Antibodies to the glycine receptor have also been described (Hutchinson et al., 2008; Mas et al., 2010). A model of the disease in mice can be produced by glutamic acid decarboxylase-specific CD4(+) T cells, and this suggests that cellular immunity may play an important role (Burton et al., 2010).
As with the stiff-person syndrome, treatment is with high doses of diazepam and baclofen. One case, with evidence of myelitis on spinal cord biopsy, improved on steroids (McCombe et al., 1989). Rituximab has also been used (Saidha et al., 2008).
Spinal alpha rigidity is a related condition resulting from isolation of spinal motor neurons from inhibitory interneuronal control (Gelfan and Tarlov, 1959). Examples have been described with trauma, cord vascular disease, cord tumors, and syringomyelia, as well as with myelitis. Most of these lesions have involved the cervical cord. Characteristically, there are stimulus-induced spasms, rigidity and abnormal limb postures involving rigid adduction, extension, and internal rotation of the affected body parts. These postures are produced by continuous motor activity which is not influenced by voluntary effort. In addition, there is wasting, weakness and loss of tendon reflexes in the arms, with long tract signs in their legs.
A variant of the stiff-person syndrome has been recognized, namely the “stiff leg” syndrome (Brown et al., 1997; Barker et al., 1998; Brown and Marsden, 1999; Fiol et al., 2001; Gurol et al., 2001; Bartsch et al., 2003). In contrast to the classic stiff-person, where continuous motor unit activity affects the back and thighs, those with the stiff-leg syndrome present with stiffness and painful spasms of one or both legs, which are rigid and dystonic. EMG findings are characteristic with continuous motor unit activity at rest, spasms of repetitive grouped discharges, and abnormal cutaneomuscular reflexes. Anti-GAD antibodies are present in about 15% of cases. Whether this is a partial syndrome or a separate disorder is debated. The prognosis is generally relatively benign with absence of other neurologic symptoms and signs for up to 16 years, but other cases can progress to the syndrome of progressive encephalomyelitis with rigidity (Gouider-Khouja et al., 2002).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


