Anterior (Carotid) Circulation
Posterior (Vertebrobasilar) Circulation
Long Circumferential Vertebrobasilar Branches
Long Penetrating Paramedian Vertebrobasilar Branches
Short Basal Vertebrobasilar Branches
Transient Ischemic Attack & Acute Ischemic Stroke
Other Causes of Intracerebral Hemorrhage
Hemorrhagic Transformation of Cerebral Infarcts
Anticoagulation & Thrombolytic Therapy
Acute Hemorrhagic Leukoencephalitis
Stroke is the fourth leading cause of death in the United States (after heart disease, cancer, and chronic lung disease) and the most common disabling neurologic disorder. Approximately 800,000 new strokes occur and approximately 130,000 people die from stroke in the United States each year.
The incidence of stroke increases with age, with approximately two-thirds of all strokes occurring in those older than 65 years. Age-adjusted stroke risk is somewhat higher in men than in women and in blacks > Hispanics > whites. Modifiable risk factors for stroke include systolic or diastolic hypertension, atrial fibrillation, diabetes, dyslipidemia, and physical inactivity (Table 13-1). The incidence of stroke has decreased in recent decades, largely because of improved treatment of hypertension, dyslipidemia, and diabetes, and reduction in smoking.
Table 13-1. Well-Documented Risk Factors for Stroke.
Nonmodifiable risk factors
Increased age
Male sex
Low birth weight
African American ethnicity
Family history of stroke
Modifiable risk factors
Vascular
Hypertension (BP >140 mm Hg systolic or 90 mm Hg diastolic)
Cigarette smoking
Asymptomatic carotid stenosis (>60% diameter)
Peripheral artery disease
Cardiac
Atrial fibrillation (with or without valvular disease)
Congestive heart failure
Coronary artery disease
Endocrine
Diabetes mellitus
Postmenopausal hormone therapy (estrogen ± progesterone)
Oral contraceptive use
Metabolic
Dyslipidemia
High total cholesterol (top 20%)
Low HDL cholesterol (<40 mg/dL)
Obesity (especially abdominal)
Hematologic
Sickle cell disease
Lifestyle
Physical inactivity
BP, blood pressure; HDL, high-density lipoprotein.
Data from Goldstein LB, et al. Guidelines for the primary prevention of stroke. A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:517-584.
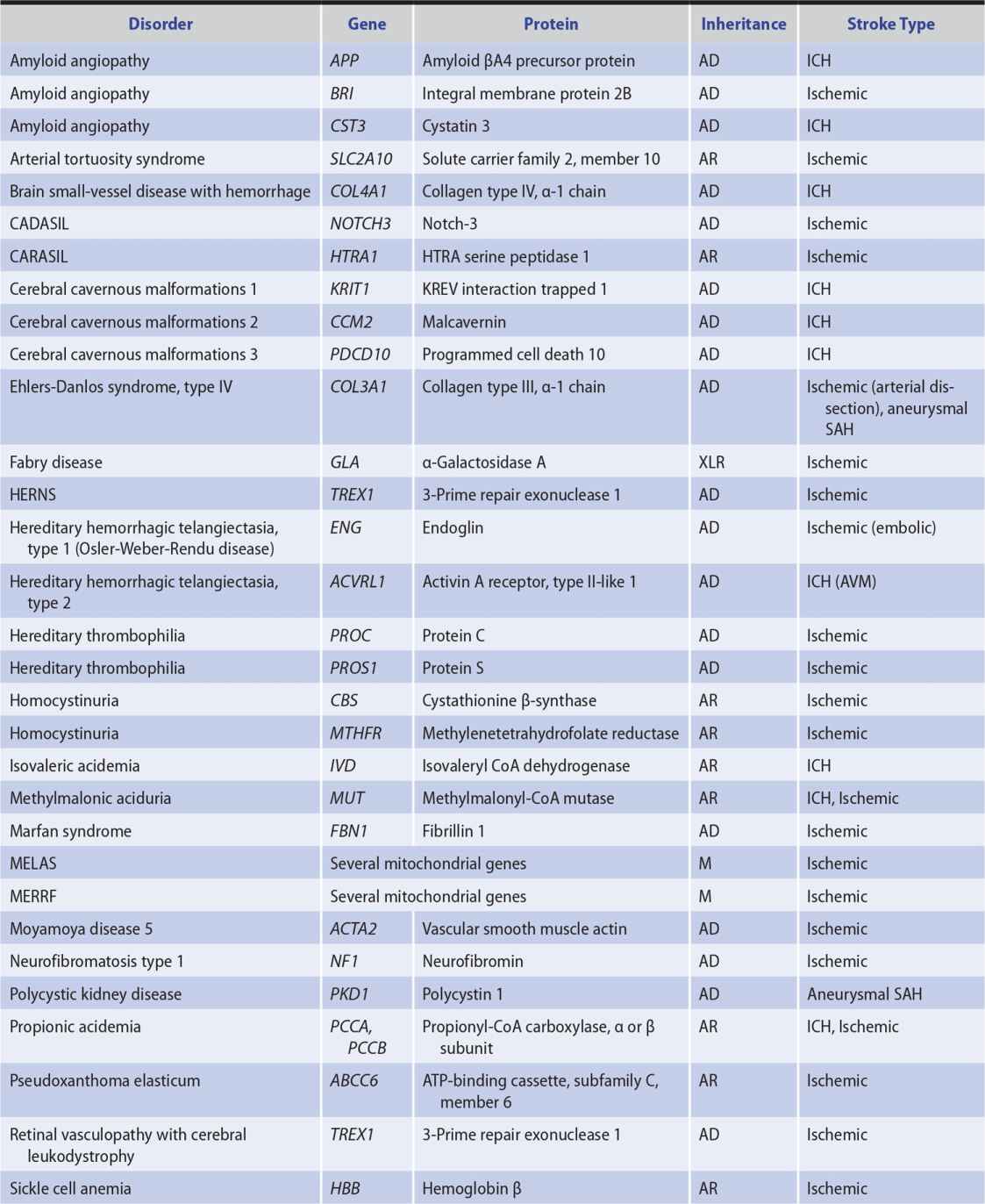
Genetic factors also appear to be important in stroke pathogenesis, although the cause of most strokes is likely to be multifactorial and involve both polygenic and environmental influences. Several, mostly rare Mendelian disorders have stroke as a major manifestation; some of these, in which the affected gene has been identified, are listed in Table 13-2.
Table 13-2. Some Monogenic Disorders Associated With Stroke.

APPROACH TO DIAGNOSIS
Stroke is a syndrome characterized by four key features:
1. Sudden onset—The sudden onset of symptoms is documented by the history.
2. Focal involvement of the central nervous system— The site of involvement is suggested by the symptoms and signs, delineated more precisely by neurologic examination, and confirmed by imaging studies (computed tomography [CT] or magnetic resonance imaging [MRI]).
3. Lack of rapid resolution—The duration of neurologic deficits is documented by the history. The classic definition of stroke required that deficits persist for at least 24 hours to distinguish stroke from transient ischemic attack (discussed later). However, any such time point is arbitrary, and transient ischemic attacks usually resolve within 1 hour. In addition, imaging studies can sometimes demonstrate prior stroke in the absence of detectable clinical deficits.
4. Vascular cause—A vascular cause may be inferred from the acute onset of symptoms and often from the patient’s age, the presence of risk factors for stroke, and the occurrence of symptoms and signs referable to the territory of a particular cerebral blood vessel. When this is confirmed by imaging studies, further investigations can be undertaken to identify a more specific etiology, such as arterial thrombosis, cardiogenic embolus, or clotting disorder.
ACUTE ONSET
Strokes begin abruptly. Neurologic deficits may be maximal at onset or may progress over seconds to hours (or occasionally days).
A stroke that is actively progressing as a direct consequence of the underlying vascular disorder (but not because of associated cerebral edema) or has done so in recent minutes is termed stroke in evolution or progressing stroke (Figure 13-1).
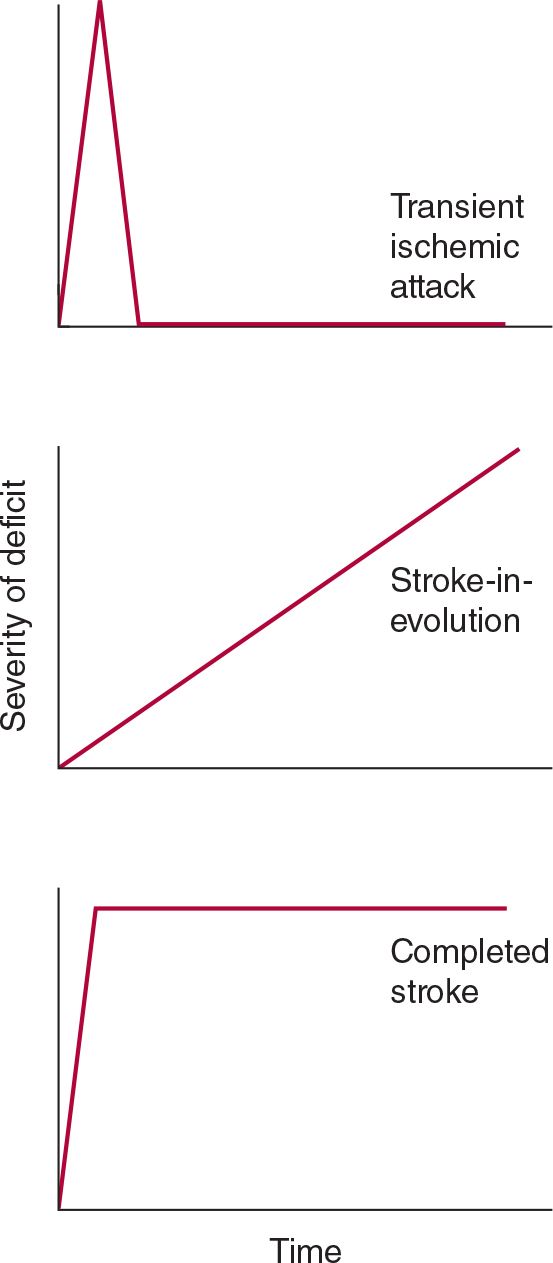
![]() Figure 13-1. Time course of cerebral ischemic events. A transient ischemic attack (TIA) produces neurologic deficits that resolve completely within a short period, usually within 1 hour. Stroke-in-evolution, or progressing stroke, causes deficits that continue to worsen even as the patient is seen. Completed stroke is defined by the presence of persistent deficits; it does not necessarily imply that the entire territory of the involved vessel is affected or that no improvement has occurred since the onset.
Figure 13-1. Time course of cerebral ischemic events. A transient ischemic attack (TIA) produces neurologic deficits that resolve completely within a short period, usually within 1 hour. Stroke-in-evolution, or progressing stroke, causes deficits that continue to worsen even as the patient is seen. Completed stroke is defined by the presence of persistent deficits; it does not necessarily imply that the entire territory of the involved vessel is affected or that no improvement has occurred since the onset.
Focal cerebral deficits that develop slowly (over weeks to months) are unlikely to be due to stroke and suggest another process, such as tumor or inflammatory or degenerative disease.
FOCAL INVOLVEMENT
Stroke produces focal symptoms and signs that correlate with the area of the brain supplied by the affected blood vessel.
In ischemic stroke, occlusion of a blood vessel interrupts the flow of blood to a specific region of the brain, interfering with neurologic functions dependent on that region and producing a more or less stereotyped pattern of deficits.
Hemorrhage produces a less predictable pattern of focal involvement because complications such as increased intracranial pressure, cerebral edema, compression of brain tissue and blood vessels, or dispersion of blood through the subarachnoid space or cerebral ventricles can impair brain function at sites remote from the hemorrhage.
Global cerebral ischemia (usually from cardiac arrest) and subarachnoid hemorrhage (discussed in Chapter 6, Headache & Facial Pain) affect the brain in more diffuse fashion and produce global cerebral dysfunction; the term stroke is not usually applied in these cases.
In most cases of stroke, the history and neurologic examination provide enough information to localize the lesion to one side of the brain (eg, to the side opposite a hemiparesis or hemisensory deficit or to the left side if aphasia is present) and to the anterior or posterior cerebral circulation.
ANTERIOR (CAROTID) CIRCULATION
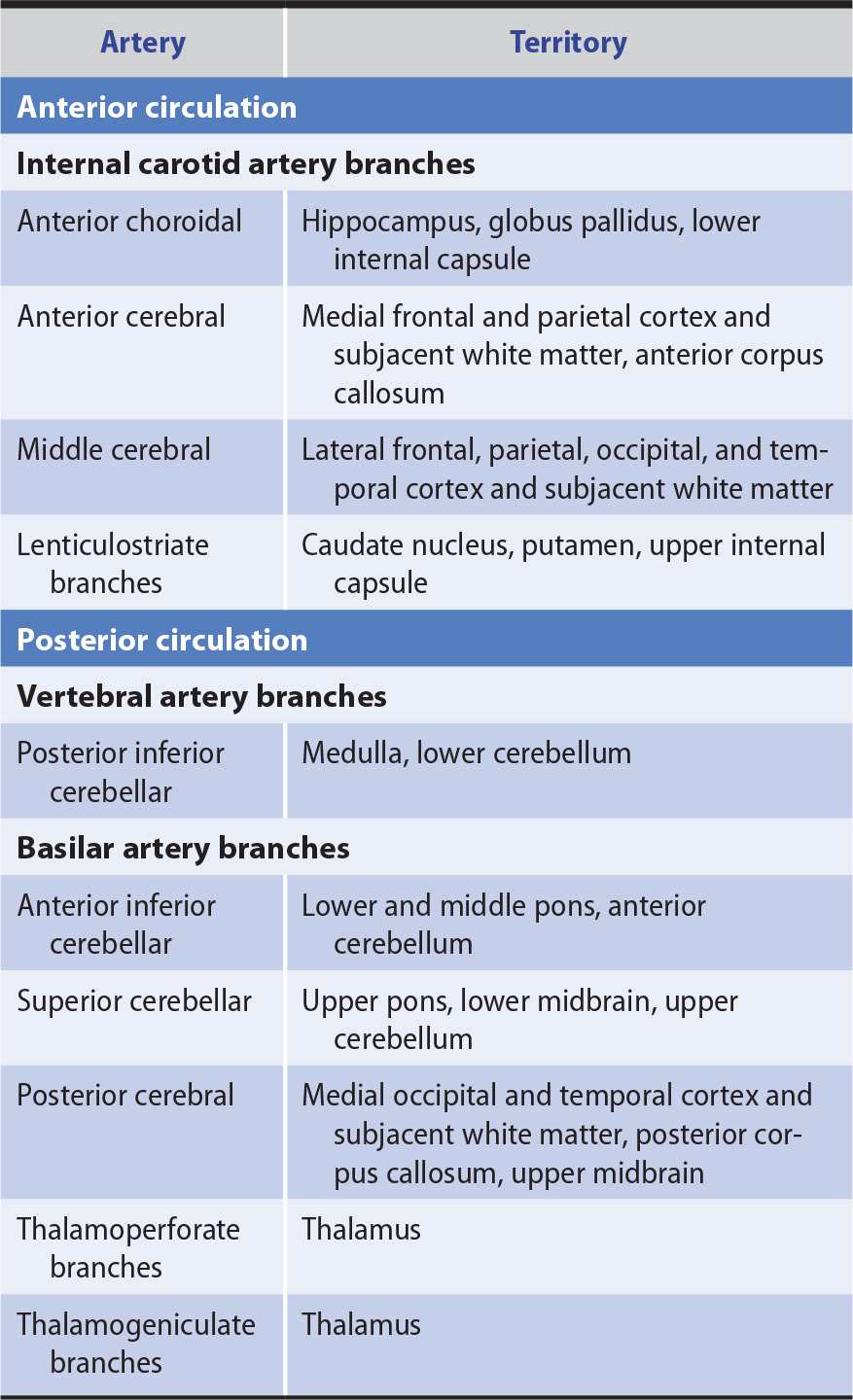
The anterior cerebral circulation supplies most of the cerebral cortex and subcortical white matter, basal ganglia, and internal capsule. It consists of the internal carotid artery and its branches: the anterior choroidal, anterior cerebral, and middle cerebral arteries. The middle cerebral artery in turn gives rise to deep, penetrating lenticulostriate branches (Figure 13-2). The specific territory of each of these vessels is listed in Table 13-3.
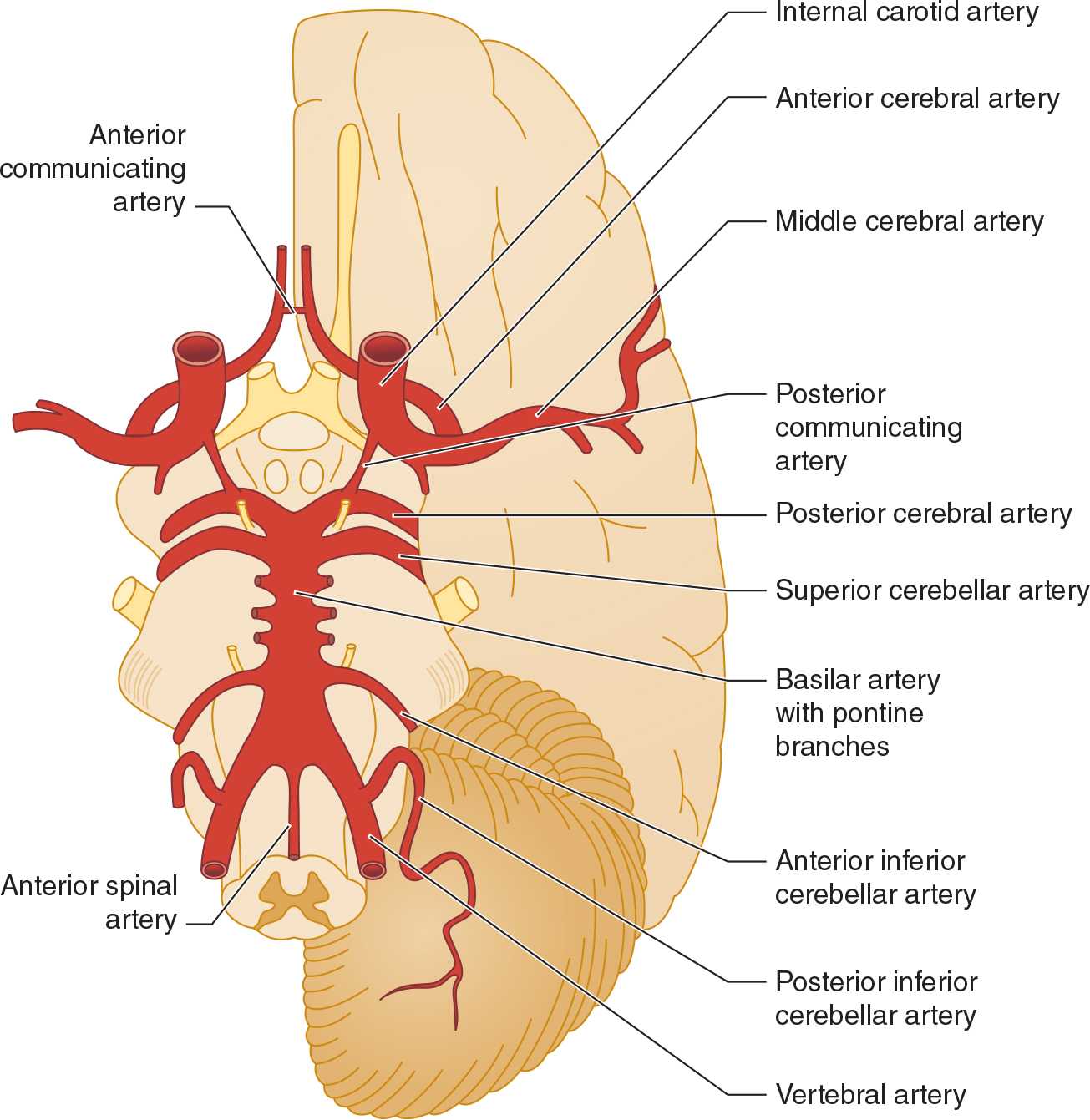
![]() Figure 13-2. Major cerebral arteries. The anterior and posterior cerebral circulations arise anterior and posterior to the posterior communicating arteries, respectively. The circle of Willis is formed by the anterior communicating, anterior cerebral, internal carotid, posterior communicating, and posterior cerebral arteries. (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
Figure 13-2. Major cerebral arteries. The anterior and posterior cerebral circulations arise anterior and posterior to the posterior communicating arteries, respectively. The circle of Willis is formed by the anterior communicating, anterior cerebral, internal carotid, posterior communicating, and posterior cerebral arteries. (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
Table 13-3. Territories of the Principal Cerebral Arteries.
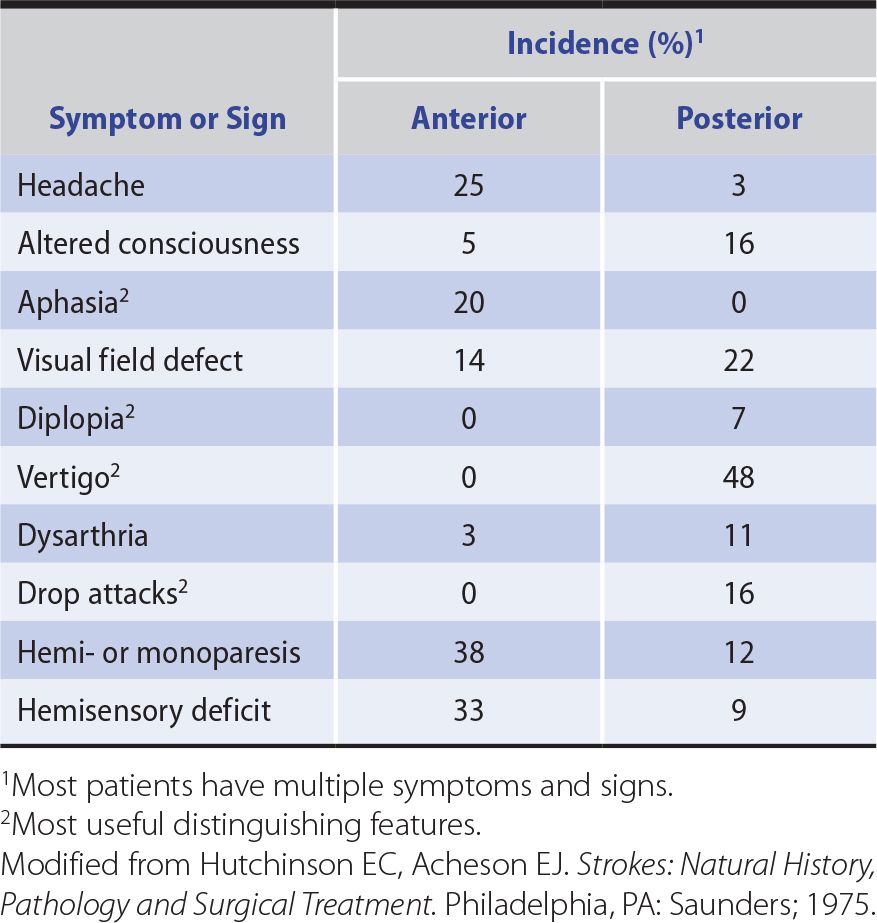
Anterior circulation strokes are commonly associated with symptoms and signs that indicate hemispheric dysfunction (Table 13-4), such as aphasia, apraxia, or agnosia (described in Chapter 1, Neurologic History & Examination). They also often produce hemiparesis, hemisensory disturbances, and visual field defects, but these can occur with posterior circulation strokes as well.
Table 13-4. Symptoms and Signs of Anterior and Posterior Circulation Ischemia.
POSTERIOR (VERTEBROBASILAR) CIRCULATION
The posterior cerebral circulation supplies the brainstem, cerebellum, thalamus, and portions of the occipital and temporal lobes. It consists of the paired vertebral arteries, the basilar artery, and their branches: the posterior inferior cerebellar, anterior inferior cerebellar, superior cerebellar, and posterior cerebral arteries (see Figure 13-2). The posterior cerebral artery also gives off thalamoperforate and thalamogeniculate branches. Areas supplied by these arteries are listed in Table 13-3.
Posterior circulation strokes produce symptoms and signs of brainstem or cerebellar dysfunction or both (see Table 13-4), including coma, drop attacks (sudden collapse without loss of consciousness), vertigo, nausea and vomiting, cranial nerve palsies, ataxia, and crossed sensorimotor deficits that affect the face on one side of the body and the limbs on the other. Hemiparesis, hemisensory disturbances, and visual field deficits also occur, but are not specific to posterior circulation strokes.
DURATION OF DEFICITS
Stroke produces neurologic deficits that persist. When symptoms and signs resolve completely after briefer periods (usually within 1 hour) without evidence of cerebral infarction, the term transient ischemic attack (TIA) is used (see Figure 13-1). Recurrent TIAs with identical clinical features (stereotypic TIAs) are usually caused by thrombosis or embolism arising from the same site within the cerebral circulation. TIAs that differ in character from event to event suggest recurrent emboli from distant (eg, cardiac) or multiple sites. Although TIAs do not themselves produce lasting neurologic dysfunction, they are important to recognize because approximately one-third of patients with TIAs will go on to have a stroke within 5 years—and because this risk may be reduced with treatment.
VASCULAR ORIGIN
Although hypoglycemia, other metabolic disturbances, trauma, and seizures can produce focal central neurologic deficits that begin abruptly and last for at least 24 hours, the term stroke is used only when such events are caused by vascular disease.
The underlying pathologic process in stroke can be either ischemia or hemorrhage, usually arising from an arterial lesion. Ischemia and hemorrhage account for approximately 90% and 10% of strokes, respectively. It may not be possible to distinguish the two by history and neurologic examination, but CT scan or MRI permits definitive diagnosis. Among ischemic strokes, about 50% are attributed to cardiac embolism, 25% to large artery occlusion, and 10% to small artery occlusion, with the remainder of unknown origin (cryptogenic).
ISCHEMIA
Interruption of blood flow to the brain deprives neurons, glia, and vascular cells of substrate glucose and oxygen. Unless blood flow is promptly restored, this leads to ischemic death of brain tissue (infarction) within the ischemic core, where flow is typically less than 20% of normal. The pattern of cell death depends on the severity of ischemia. With mild ischemia, as in cardiac arrest with rapid reperfusion, selective vulnerability of certain neuronal populations may be observed. More severe ischemia produces selective neuronal necrosis, in which most or all neurons die but glia and vascular cells are preserved. Complete, permanent ischemia, such as occurs in stroke without reperfusion, causes pannecrosis, affecting all cell types, resulting in chronic cavitary lesions.
Where ischemia is incomplete (20%-40% of normal blood flow)—as in the ischemic border zone or penumbra—cell damage is potentially reversible and cell survival may be prolonged. However, unless blood flow is restored, by recanalization of the occluded vessel or collateral circulation from other vessels, reversibly damaged cells begin to die as well, and the infarct expands. Death of penumbral tissue is associated with a worse clinical outcome.
Brain edema is another determinant of stroke outcome. Ischemia leads to vasogenic edema as fluid leaks from the intravascular compartment into brain parenchyma. Edema is usually maximal approximately 2 to 3 days after stroke and may be sufficiently severe that it produces a mass effect that causes herniation (displacement of brain tissue between intracranial compartments) and death.
Two pathogenetic mechanisms can produce ischemic stroke—thrombosis and embolism. However, the distinction is often difficult or impossible to make on clinical grounds.
 Thrombosis
Thrombosis
Thrombosis produces stroke by occluding large cerebral arteries (especially the internal carotid, middle cerebral, or basilar), small penetrating arteries (as in lacunar stroke), cerebral veins, or venous sinuses. Symptoms typically evolve over minutes to hours. Thrombotic strokes are often preceded by TIAs, which tend to produce similar symptoms because they affect the same territory recurrently.
 Embolism
Embolism
Embolism produces stroke when cerebral arteries are occluded by the distal passage of thrombus from the heart, aortic arch, or large cerebral arteries. Emboli in the anterior cerebral circulation most often occlude the middle cerebral artery or its branches, because most hemispheric blood flow is through this vessel. Emboli in the posterior cerebral circulation usually lodge at the apex of the basilar artery or in the posterior cerebral arteries. Embolic strokes often produce neurologic deficits that are maximal at onset. When TIAs precede embolic strokes, especially those arising from a cardiac source, symptoms typically vary between attacks because different vascular territories are affected.
HEMORRHAGE
Hemorrhage may interfere with cerebral function through a variety of mechanisms, including destruction or compression of brain tissue, compression of vascular structures, and edema. Intracranial hemorrhage is classified by its location as intracerebral, subarachnoid, subdural, or epidural, all of which—except subdural hemorrhage—are usually caused by arterial bleeding.
 Intracerebral Hemorrhage
Intracerebral Hemorrhage
Intracerebral hemorrhage causes symptoms by destroying or compressing brain tissue. Unlike ischemic stroke, intracerebral hemorrhage tends to cause more severe headache and depression of consciousness as well as neurologic deficits that do not correspond to the distribution of any single blood vessel.
 Subarachnoid Hemorrhage
Subarachnoid Hemorrhage
Subarachnoid hemorrhage leads to cerebral dysfunction due to increased intracranial pressure, resulting hypoperfusion, direct destruction of tissue, and toxic constituents of subarachnoid blood. Subarachnoid hemorrhage may be complicated by vasospasm (leading to ischemia), rebleeding, extension of blood into brain tissue (producing an intracerebral hematoma), or hydrocephalus. Subarachnoid hemorrhage typically presents with headache rather than focal neurologic deficits; it is discussed in Chapter 6, Headache & Facial Pain.
 Subdural or Epidural Hemorrhage
Subdural or Epidural Hemorrhage
Subdural or epidural hemorrhage produces a mass lesion that can compress the underlying brain. These hemorrhages are often traumatic in origin and usually present with headache or altered consciousness. Because of their importance as causes of coma, subdural and epidural hemorrhages are discussed in Chapter 3, Coma.
FOCAL CEREBRAL ISCHEMIA
PATHOPHYSIOLOGY
The pathophysiology of focal cerebral ischemia is complex, as it involves a process that evolves over time, affects the brain nonuniformly, and targets multiple cell types. Nevertheless, several potentially important underlying mechanisms have been identified. Some mechanisms are likely to have their major impact early and others later in the course of stroke. Moreover, some contribute to ischemic injury, whereas others promote tissue survival or repair.
INJURY MECHANISMS
 Energy Failure
Energy Failure
Neurons rely on oxidative metabolism to generate adenosine triphosphate (ATP) for their high energy demands. Reduction of blood flow interferes with the delivery of two key substrates for this process—oxygen and glucose—causing ATP levels to fall. Cells can compensate to a limited extent by generating ATP via glycolysis but, without prompt reperfusion, they cease to function and eventually die. Like other ischemic injury mechanisms, energy failure is most pronounced in the ischemic core and less so in the surrounding penumbra.
 Ion Gradients
Ion Gradients
A major use of cellular energy is the maintenance of transmembrane ion gradients. With energy failure, these are dissipated. Na+/K+-ATPase, which accounts for the majority of neuronal energy expenditure and is responsible for maintaining high intracellular K+ concentrations, fails to do so. K+ leaks from cells and depolarizes adjacent cells, activating voltage-gated ion channels and neurotransmitter release. Extracellular K+ and neurotransmitter glutamate trigger cortical spreading depression, leading to further neuron and astrocyte depolarization. This consumes additional energy and may extend the infarct.
 Calcium Dysregulation
Calcium Dysregulation
Ca2+ is maintained at low resting cytoplasmic levels, but ischemic elevation of extracellular K+ causes membrane depolarization and triggers Ca2+ entry. Catabolic proteases, lipases, and nucleases are activated, mitochondrial function is compromised, and cell death pathways are mobilized.
 Excitotoxicity
Excitotoxicity
Excitotoxicity refers to the neurotoxic effects of excitatory neurotransmitters, especially glutamate. Ischemia promotes excitotoxicity by stimulating neuronal glutamate release, reversing astrocytic glutamate uptake, and activating glutamate receptor-coupled ion channels. Influx of Ca2+ through these channels contributes to Ca2+ dysregulation and activates neuronal nitric oxide synthase, generating potentially neurotoxic nitric oxide.
 Oxidative and Nitrosative Injury
Oxidative and Nitrosative Injury
Some toxic effects of ischemia are mediated by highly reactive oxidative and nitrosative compounds including superoxide and nitric oxide, which act primarily during the reperfusion phase that follows ischemia. Their effects include inhibiting mitochondrial enzymes and function, damaging DNA, activating ion channels, causing covalent modification of proteins, and triggering cell death pathways.
 Cell Death Cascades
Cell Death Cascades
Ischemic cell death occurs most rapidly in the infarct core and more slowly in the penumbra and during reperfusion. Rapid cell death involves necrosis, in which cells and organelles swell, membranes rupture, and cellular contents spill into the extracellular space, whereas more delayed (programmed) cell death (eg, apoptosis) predominates in the penumbra and during reperfusion.
 Inflammation
Inflammation
Cerebral ischemia triggers an inflammatory response that involves both resident and blood-borne cells of the innate immune system. The former include astrocytes and microglia, and the latter neutrophils, lymphocytes, and monocytes. Adaptive immune responses emerge later in the course. Molecular mediators of ischemia-induced inflammation include adhesion molecules, cytokines, chemokines, and proteases. Although the early inflammatory response to ischemia exacerbates injury, subsequent inflammatory events may be neuroprotective or contribute to repair.
SURVIVAL & REPAIR MECHANISMS
 Collateral Circulation
Collateral Circulation
The first line of defense against ischemia is collateral circulation, which, if adequate, can bypass an arterial occlusion. The cerebral circulation contains numerous collateral pathways, accounting for the observation that patients with total occlusion of a major vessel are sometimes asymptomatic. However, this is not always the case, especially when occlusion is abrupt. Examples of collateral routes for cerebral blood flow include the following:
1. Bilateral vertebral artery occlusion—anterior spinal artery
2. Common carotid artery occlusion—contralateral common carotid artery via ipsilateral external carotid artery or vertebral artery via ipsilateral occipital artery
3. Internal carotid artery occlusion—ipsilateral external carotid artery via ophthalmic artery or circle of Willis
4. Middle cerebral artery occlusion—ipsilateral anterior or posterior cerebral artery via leptomeningeal anastomoses
 Inhibitory Neurotransmitters
Inhibitory Neurotransmitters
Enhanced tonic inhibition mediated through extrasynaptic GABAA receptors may mitigate excitotoxic injury early in the course of stroke. However, persistent inhibition may impair recovery.
 Transcriptional Hypoxia Response
Transcriptional Hypoxia Response
Hypoxia activates transcription of proteins that promote cell survival and tissue recovery, including glycolytic enzymes, erythropoietin, and vascular endothelial growth factor. Other cytoprotective proteins induced after ischemia include antiapoptotic proteins, growth factors, and heat-shock proteins.
 Neurogenesis
Neurogenesis
Cerebral ischemia stimulates neurogenesis and some new neurons migrate to ischemic brain regions. Here they may promote survival and repair by releasing growth factors, suppressing inflammation, or other effects.
 Angiogenesis
Angiogenesis
Ischemia also stimulates capillary sprouting to enhance local blood supply. The impact of this process (angiogenesis) in the acute phase of stroke is uncertain, but it may help to protect against subsequent ischemic episodes.
 Ischemic Tolerance
Ischemic Tolerance
Ischemia may provide paradoxical protection against subsequent ischemia through ischemic tolerance, in which mild ischemia preconditions brain tissue and confers relative ischemia resistance. Ischemic tolerance involves extensive changes in gene expression and numerous molecular mediators.
 Repair Mechanisms
Repair Mechanisms
Most patients recover to some extent after stroke, reflecting a capacity for spontaneous postischemic repair and the brain’s innate plasticity. Plastic changes occur in the peri-infarct region and at remote sites, such as the contralateral cerebral hemisphere, and include changes in gene expression, increased neuronal excitability, axonal sprouting, synaptogenesis, somatotopic reorganization, and formation of new neuronal circuits.
PATHOLOGY
LARGE ARTERY OCCLUSION
On gross inspection, a recent infarct from large artery occlusion is a swollen, softened area of brain that usually affects both gray and white matter. Microscopy shows acute ischemic changes in neurons (shrinkage, microvacuolization, dark staining), destruction of glial cells, necrosis of small blood vessels, disruption of nerve axons and myelin, and accumulation of interstitial fluid. Perivascular hemorrhages may be observed. Depending on the interval between infarction and death, cerebral edema may also be present. It is maximal during the first 4 or 5 days after stroke and can cause herniation of the cingulate gyrus across the midline or of the temporal lobe below the tentorium (see Figure 3-4 in Chapter 3, Coma).
SMALL ARTERY OCCLUSION
Infarcts from small artery occlusion rarely cause death, so only chronic lesions are usually found at autopsy. These include lacunes, or small cavities up to ~15 mm in diameter usually located in subcortical white (eg, internal capsule) or deep gray (eg, basal ganglia or thalamus) matter; white matter (including periventricular) lesions showing punctate or confluent myelin rarefaction, gliosis, and axonal loss; and microbleeds. Small vessel occlusion may be associated with atherosclerosis, lipohyalinosis (collagenous thickening and inflammation of the vessel wall), or fibrinoid necrosis (vessel-wall destruction with perivascular inflammation).
CLINICAL-ANATOMIC CORRELATION
Infarction in the distribution of different cerebral arteries often produces distinctive clinical syndromes, which can facilitate anatomic and etiologic diagnosis and help guide treatment.
ANTERIOR CEREBRAL ARTERY
 Anatomy
Anatomy
The anterior cerebral artery supplies the parasagittal cerebral cortex (Figures 13-3 and 13-4), which includes portions of motor and sensory cortex related to the contralateral leg, the so-called bladder inhibitory or micturition center, and the anterior corpus callosum.
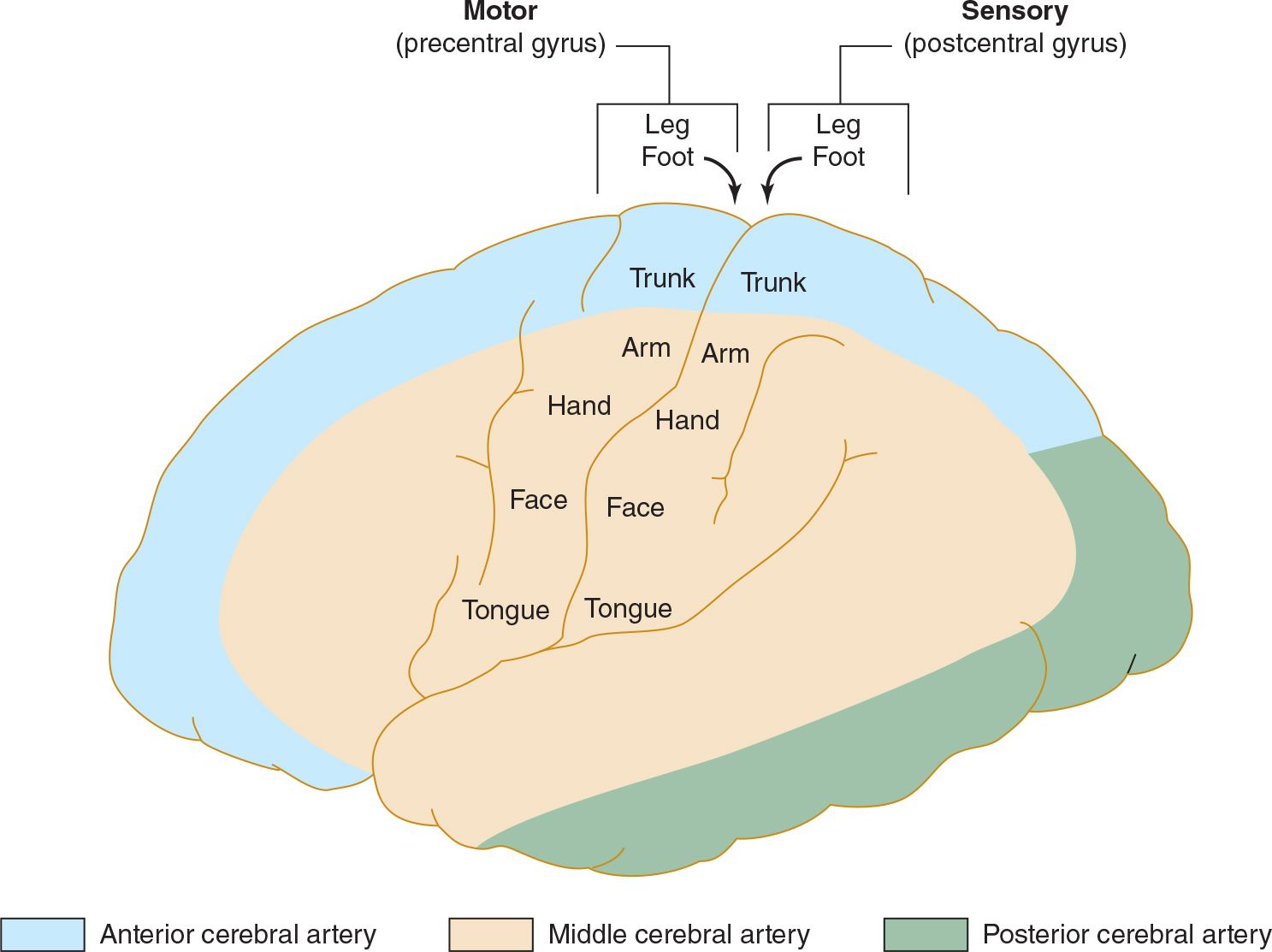
![]() Figure 13-3. Arterial supply of the primary motor and sensory cortex (lateral view). The middle cerebral artery supplies areas of the primary motor and sensory cortex related to face and arm function, whereas the anterior cerebral artery supplies areas related to leg function. This explains why middle cerebral artery strokes affect the face and arm most severely, whereas anterior cerebral artery strokes affect the leg. (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
Figure 13-3. Arterial supply of the primary motor and sensory cortex (lateral view). The middle cerebral artery supplies areas of the primary motor and sensory cortex related to face and arm function, whereas the anterior cerebral artery supplies areas related to leg function. This explains why middle cerebral artery strokes affect the face and arm most severely, whereas anterior cerebral artery strokes affect the leg. (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
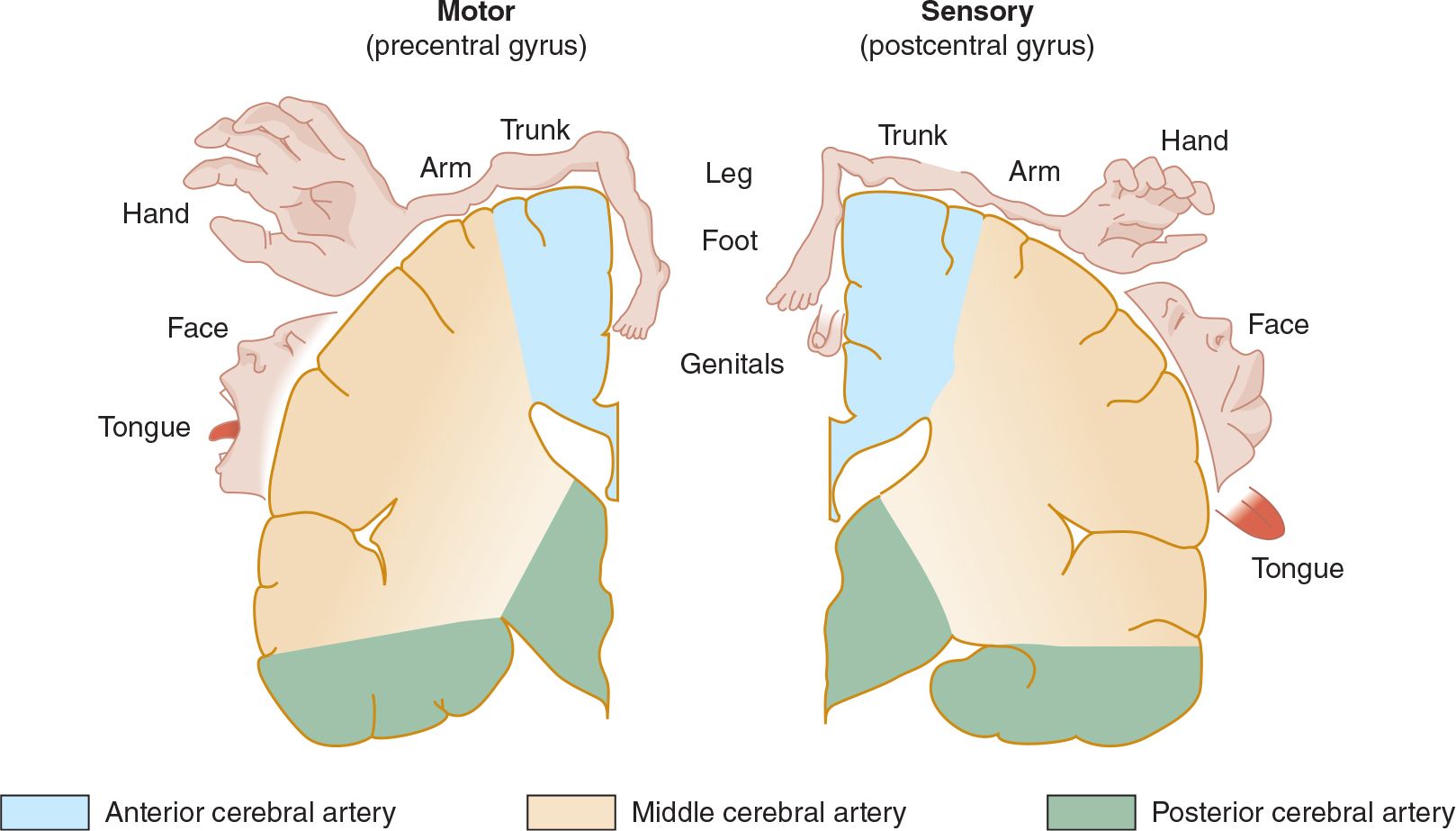
![]() Figure 13-4. Arterial supply of the primary motor and sensory cortex (coronal view). (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
Figure 13-4. Arterial supply of the primary motor and sensory cortex (coronal view). (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
 Clinical Syndrome
Clinical Syndrome
Anterior cerebral artery strokes produce contralateral paralysis and sensory loss exclusively or primarily affecting the leg. There may also be abulia (apathy), disconnection syndromes such as the alien hand (involuntary performance of complex motor activity), transcortical expressive aphasia (see Table 1-1 in Chapter 1, Neurologic History & Examination), and urinary incontinence.
MIDDLE CEREBRAL ARTERY
 Anatomy
Anatomy
The middle cerebral artery supplies most of the remainder of the cerebral hemisphere and deep subcortical structures (see Figures 13-3 and 13-4). Cortical branches include the superior division, which supplies motor and sensory representation of the face, hand, and arm, and the expressive language (Broca) area of the dominant hemisphere (Figure 13-5). The inferior division supplies the visual radiations, visual cortex related to macular vision, and the receptive language (Wernicke) area of the dominant hemisphere. Lenticulostriate branches of the most proximal portion (stem) of the middle cerebral artery supply the basal ganglia and motor fibers to the face, hand, arm, and leg as they descend in the genu and the posterior limb of the internal capsule.
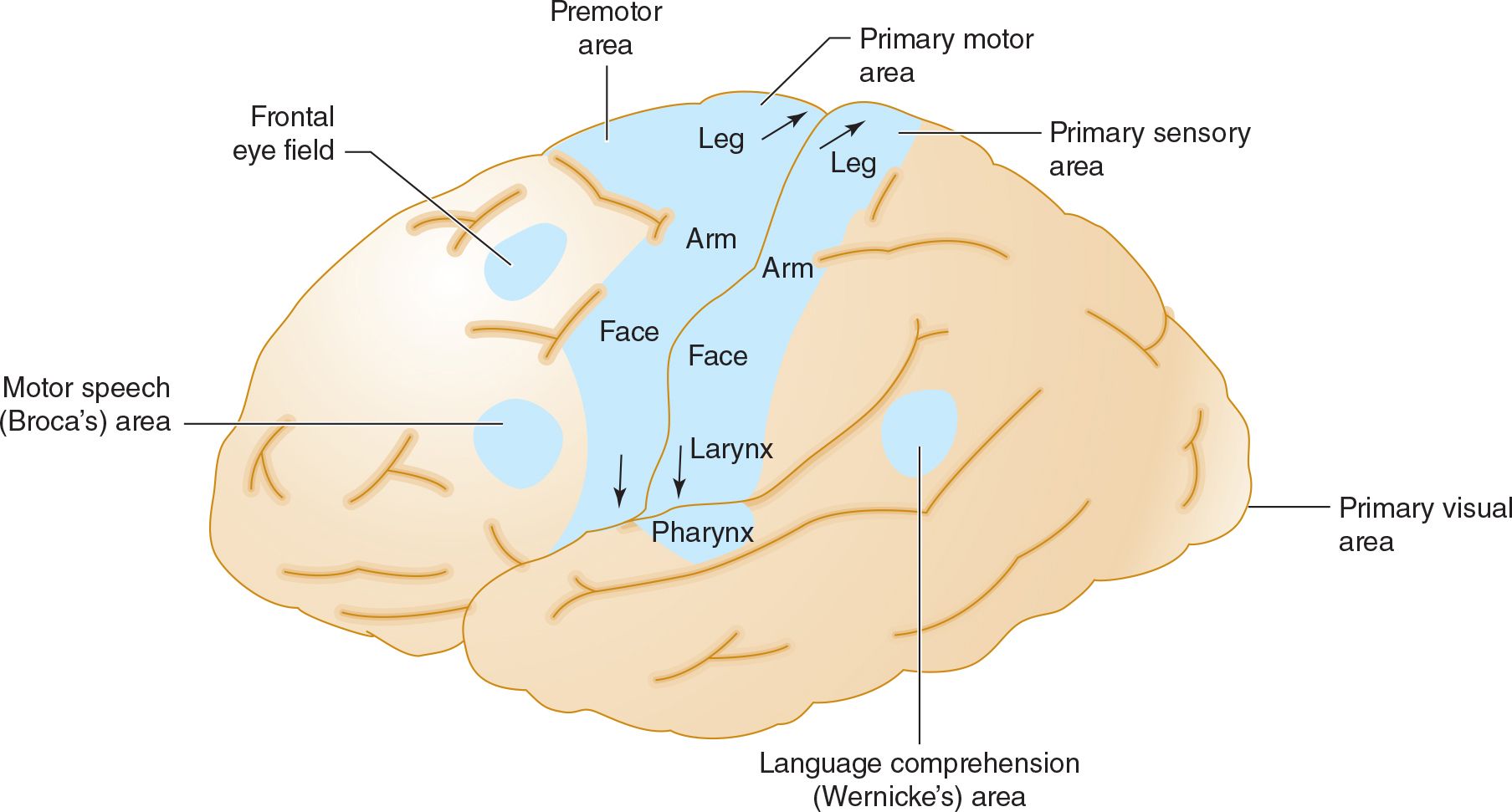
![]() Figure 13-5. Anatomic basis of middle cerebral artery syndromes. Stroke in the distribution of the middle cerebral artery causes hemiparesis affecting primarily face and arm (due to involvement of the primary motor area), hemisensory deficit affecting primarily face and arm (due to involvement of the primary sensory area), gaze preference toward the affected hemisphere (due to involvement of the frontal eye field), aphasia if the dominant hemisphere is affected (due to involvement of Broca area, Wernicke area, or both), and hemianopia (due to involvement of the optic radiations leading to the primary visual area. (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
Figure 13-5. Anatomic basis of middle cerebral artery syndromes. Stroke in the distribution of the middle cerebral artery causes hemiparesis affecting primarily face and arm (due to involvement of the primary motor area), hemisensory deficit affecting primarily face and arm (due to involvement of the primary sensory area), gaze preference toward the affected hemisphere (due to involvement of the frontal eye field), aphasia if the dominant hemisphere is affected (due to involvement of Broca area, Wernicke area, or both), and hemianopia (due to involvement of the optic radiations leading to the primary visual area. (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
 Clinical Syndrome
Clinical Syndrome
Depending on the site of involvement, several clinical syndromes can occur.
1. Superior division stroke results in contralateral hemiparesis that affects the face, hand, and arm but spares the leg, and contralateral hemisensory deficit in the same distribution, but no homonymous hemianopia. If the dominant hemisphere is involved, there is Broca (expressive) aphasia, which is characterized by impaired language expression with intact comprehension.
2. Inferior division stroke results in contralateral homonymous hemianopia that may be denser inferiorly, impaired cortical sensory functions (eg, graphesthesia and stereognosis) on the contralateral side of the body, and disorders of spatial thought (eg, anosognosia [unawareness of deficit], neglect of the contralateral limbs and contralateral side of external space, dressing apraxia, and constructional apraxia). If the dominant hemisphere is involved, Wernicke (receptive) aphasia occurs and is manifested by impaired comprehension and fluent but often nonsensical speech. With involvement of the nondominant hemisphere, an acute confusional state may occur.
3. Occlusion at the bifurcation or trifurcation of the middle cerebral artery combines the features of superior and inferior division stroke, including contralateral hemiparesis and hemisensory deficit involving the face and arm more than leg, homonymous hemianopia, and—if the dominant hemisphere is affected—global (combined expressive and receptive) aphasia.
4. Occlusion of the stem of the middle cerebral artery occurs proximal to the origin of the lenticulostriate branches, resulting in a clinical syndrome similar to that seen after occlusion at the trifurcation. In addition, however, involvement of the internal capsule causes paralysis of the contralateral leg, so hemiplegia and sensory loss affect face, hand, arm, and leg.
INTERNAL CAROTID ARTERY
 Anatomy
Anatomy
The internal carotid artery arises at the bifurcation of the common carotid artery in the neck. In addition to the anterior and middle cerebral arteries, it also gives rise to the ophthalmic artery, which supplies the retina.
 Clinical Syndrome
Clinical Syndrome
Internal carotid artery occlusion may be asymptomatic, or cause strokes of highly variable severity, depending on the adequacy of collateral circulation. Symptomatic occlusion results in a syndrome similar to that of middle cerebral artery stroke (contralateral hemiplegia, hemisensory deficit, and homonymous hemianopia, together with aphasia if the dominant hemisphere is involved). Monocular blindness is also common.
POSTERIOR CEREBRAL ARTERY
 Anatomy
Anatomy
The paired posterior cerebral arteries arise from the tip of the basilar artery in most cases (Figure 13-6) and supply the occipital cerebral cortex, medial temporal lobes, posterior corpus callosum, thalamus, and rostral midbrain. Emboli in the basilar artery tend to lodge at its apex and occlude one or both posterior cerebral arteries; subsequent fragmentation can produce asymmetric or patchy posterior cerebral artery infarction.
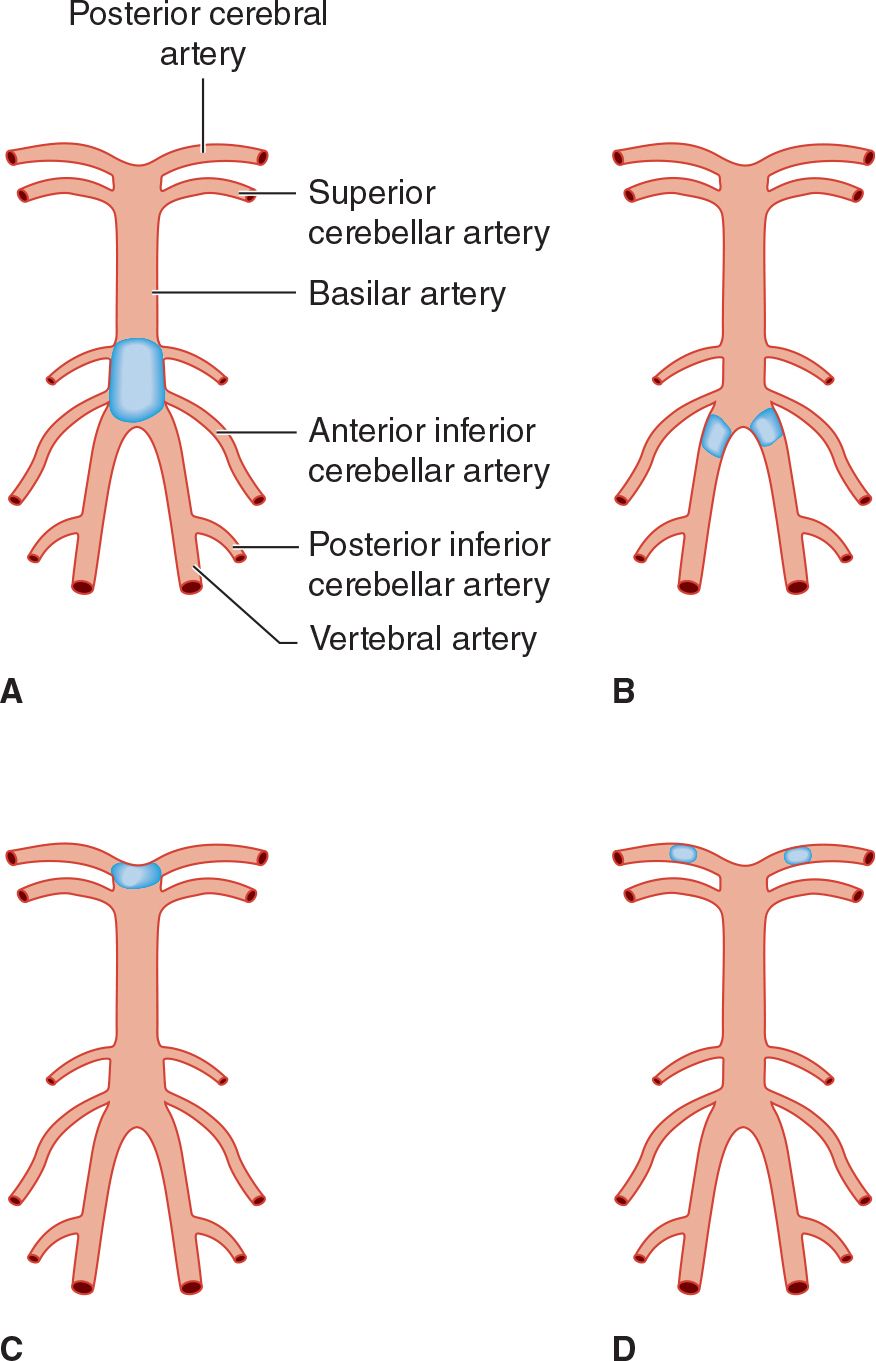
![]() Figure 13-6. Sites of thrombotic and embolic occlusions in the vertebrobasilar circulation. (A) Thrombotic occlusion of the basilar artery. (B) Thrombotic occlusion of both vertebral arteries. (C) Embolic occlusion at the apex of the basilar artery. (D) Embolic occlusion of both posterior cerebral arteries.
Figure 13-6. Sites of thrombotic and embolic occlusions in the vertebrobasilar circulation. (A) Thrombotic occlusion of the basilar artery. (B) Thrombotic occlusion of both vertebral arteries. (C) Embolic occlusion at the apex of the basilar artery. (D) Embolic occlusion of both posterior cerebral arteries.
 Clinical Syndrome
Clinical Syndrome
Posterior cerebral artery occlusion produces homonymous hemianopia affecting the contralateral visual field, except that macular vision may be spared. In contrast to visual field defects from infarction in the middle cerebral artery territory, those caused by posterior cerebral artery occlusion may be denser superiorly. With occlusion near the origin of the posterior cerebral artery at the level of the midbrain, ocular abnormalities may occur, including vertical gaze palsy, oculomotor (III) nerve palsy, internuclear ophthalmoplegia, and vertical skew deviation of the eyes. Involvement of the occipital lobe of the dominant hemisphere may cause anomic aphasia (difficulty in naming objects), alexia without agraphia (inability to read, with no impairment of writing), or visual agnosia. The last is failure to identify objects presented in the left side of the visual field, caused by a lesion of the corpus callosum that disconnects the right visual cortex from language areas of the left hemisphere. Bilateral posterior cerebral artery infarction may result in cortical blindness, memory impairment (from temporal lobe involvement), or inability to recognize familiar faces (prosopagnosia), as well as a variety of exotic visual and behavioral syndromes.
BASILAR ARTERY
 Anatomy
Anatomy
The basilar artery arises from the junction of the paired vertebral arteries (see Figure 13-6) and courses over the ventral surface of the brainstem to terminate at the level of the midbrain, where it bifurcates to form the posterior cerebral arteries. Branches of the basilar artery supply the occipital and medial temporal lobes, medial thalamus, posterior limb of the internal capsule, brainstem, and cerebellum.
 Clinical Syndromes
Clinical Syndromes
1. Thrombosis—Thrombotic occlusion of the basilar artery or both vertebral arteries (see Figure 13-6) is often incompatible with survival. It causes bilateral symptoms and signs of brainstem and cerebellar dysfunction from involvement of multiple branch arteries (Figure 13-7). Temporary occlusion of one or both vertebral arteries can also result from rotating the head in patients with cervical spondylosis, leading to transient brainstem dysfunction.
Stenosis or occlusion of the subclavian artery before it has given rise to the vertebral artery can lead to the subclavian steal syndrome, in which blood passes from the vertebral artery into the distal subclavian artery with physical activity of the ipsilateral arm. The syndrome is not predictive of stroke in the vertebrobasilar system.
Basilar thrombosis usually affects the proximal basilar artery (see Figure 13-6), which supplies the pons. Involvement of the dorsal pons (tegmentum) produces unilateral or bilateral abducens (VI) nerve palsy; horizontal eye movements are impaired, but vertical nystagmus and ocular bobbing may be present. The pupils are constricted due to involvement of descending sympathetic pupillodilator fibers, but may be reactive. Hemiplegia or quadriplegia is usually present, and coma is common. A CT or MRI brain scan will differentiate between basilar occlusion and pontine hemorrhage.
In some patients, the ventral pons (basis pontis) is infarcted and the tegmentum is spared. Such patients remain conscious but quadriplegic (locked-in syndrome). Locked-in patients may be able to open or move their eyes vertically on command. A normal conventional electroencephalogram (EEG) further distinguishes the locked-in state from coma (see Chapter 3, Coma).
2. Embolism—Emboli in the basilar artery usually lodge at its apex (see Figure 13-6). Interruption of blood flow to the ascending reticular formation in the midbrain and thalamus produces immediate loss or impairment of consciousness. Unilateral or bilateral oculomotor (III) nerve palsies are characteristic. Hemiplegia or quadriplegia with decerebrate or decorticate posturing results from involvement of the cerebral peduncles in the midbrain. Thus the top of the basilar syndrome may be confused with midbrain damage caused by transtentorial uncal herniation. Less commonly, an embolus may lodge more proximally, producing a syndrome indistinguishable from basilar thrombosis.
Smaller emboli may occlude the rostral basilar artery transiently before fragmenting and passing into one or both posterior cerebral arteries (see Figure 13-6). In such cases, portions of the midbrain, thalamus, and temporal and occipital lobes can be infarcted. Patients may display visual (homonymous hemianopia, cortical blindness), visuomotor (impaired convergence, paralysis of upward or downward gaze, diplopia), and behavioral (especially confusion) abnormalities without prominent motor dysfunction. Sluggish pupillary responses are a helpful sign of midbrain involvement.
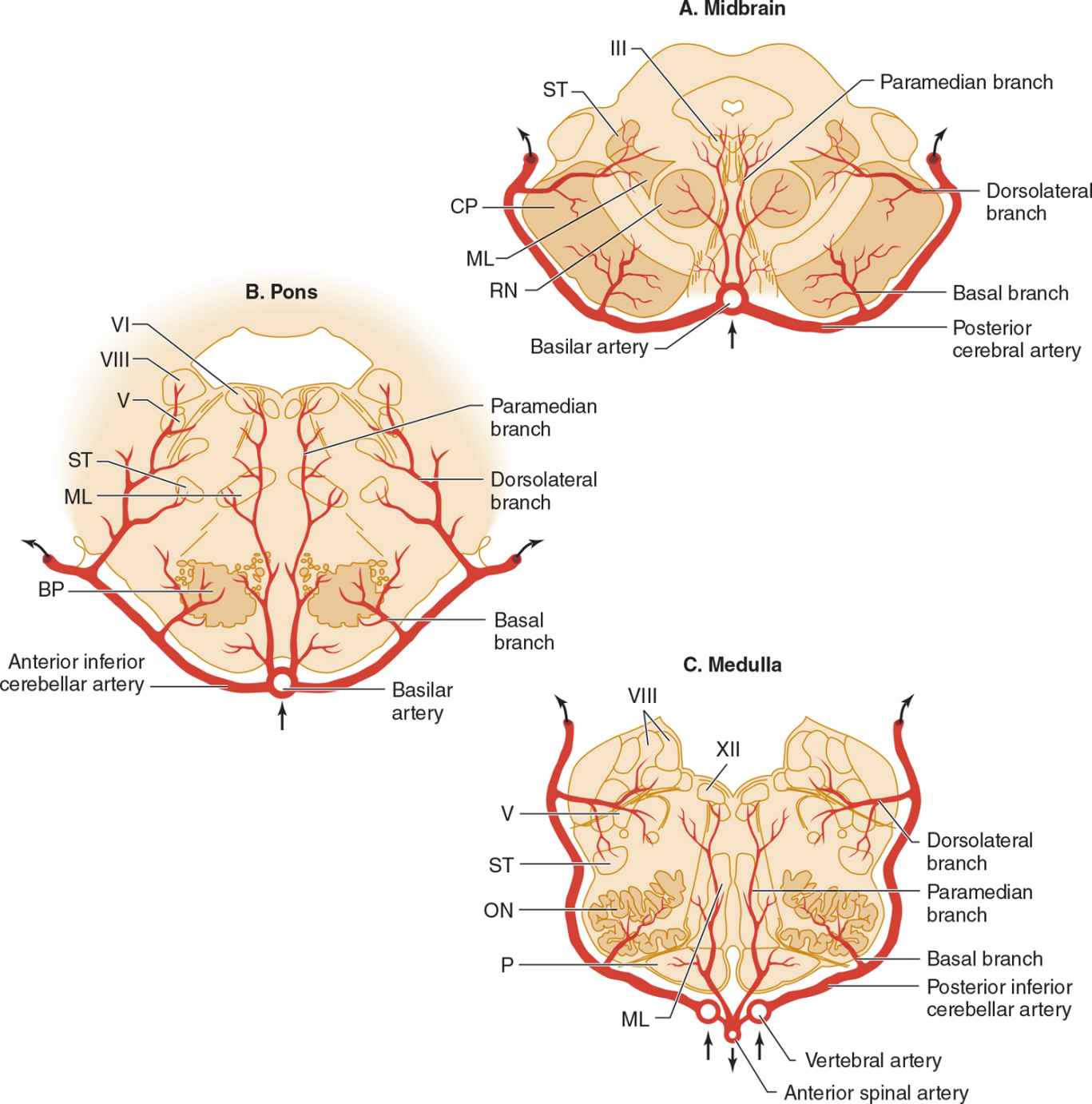
![]() Figure 13-7. Arterial supply of the brainstem. (A) Midbrain. The basilar artery gives off paramedian branches that supply the oculomotor (III) nerve nucleus and red nucleus (RN). A larger branch, the posterior cerebral artery, courses laterally around the midbrain, giving off a basal branch that supplies the cerebral peduncle (CP) and a dorsolateral branch supplying the spinothalamic tract (ST) and medial lemniscus (ML). The posterior cerebral artery continues (upper arrows) to supply the thalamus, occipital lobe, and medial temporal lobe. (B) Pons. Paramedian branches of the basilar artery supply the abducens (VI) nucleus and medial lemniscus (ML). The anterior inferior cerebellar artery gives off a basal branch to descending motor pathways in the basis pontis (BP) and a dorsolateral branch to the trigeminal (V) nucleus, vestibular (VIII) nucleus, and spinothalamic tract (ST), before passing to the cerebellum (upper arrows). (C) Medulla. Paramedian branches of the vertebral arteries supply descending motor pathways in the pyramid (P), the medial lemniscus (ML), and the hypoglossal (XII) nucleus. Another branch, the posterior inferior cerebellar artery, gives off a basal branch to the olivary nuclei (ON) and a dorsolateral branch that supplies the trigeminal (V) nucleus, vestibular (VIII) nucleus, and spinothalamic tract (ST), on its way to the cerebellum (upper arrows). (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)
Figure 13-7. Arterial supply of the brainstem. (A) Midbrain. The basilar artery gives off paramedian branches that supply the oculomotor (III) nerve nucleus and red nucleus (RN). A larger branch, the posterior cerebral artery, courses laterally around the midbrain, giving off a basal branch that supplies the cerebral peduncle (CP) and a dorsolateral branch supplying the spinothalamic tract (ST) and medial lemniscus (ML). The posterior cerebral artery continues (upper arrows) to supply the thalamus, occipital lobe, and medial temporal lobe. (B) Pons. Paramedian branches of the basilar artery supply the abducens (VI) nucleus and medial lemniscus (ML). The anterior inferior cerebellar artery gives off a basal branch to descending motor pathways in the basis pontis (BP) and a dorsolateral branch to the trigeminal (V) nucleus, vestibular (VIII) nucleus, and spinothalamic tract (ST), before passing to the cerebellum (upper arrows). (C) Medulla. Paramedian branches of the vertebral arteries supply descending motor pathways in the pyramid (P), the medial lemniscus (ML), and the hypoglossal (XII) nucleus. Another branch, the posterior inferior cerebellar artery, gives off a basal branch to the olivary nuclei (ON) and a dorsolateral branch that supplies the trigeminal (V) nucleus, vestibular (VIII) nucleus, and spinothalamic tract (ST), on its way to the cerebellum (upper arrows). (Used with permission from Waxman S. Clinical Neuroanatomy. 26th ed. New York, NY: McGraw-Hill; 2010.)




