Synaptic Plasticity
Christophe Bernard
Introduction
Cognitive tasks are associated with specific firing patterns of neurons.27,60 During learning and memory processes, a conditioning stimulus can trigger morphological and modifications at the neuronal network level to store novel information. This is known as synaptic plasticity. These modifications can result in changes in firing patterns in response to the same unconditioned stimulus. For example, in the cerebellum, the vestibulo-ocular reflex can be adaptively modified when vestibular-visual conditions are changed, with the triggering error signal being provided by the firing of climbing fibers.52 The goal of this chapter is to describe the various mechanisms that have been proposed to explain how information processing can be modified via dynamic modifications at the synapse. These mechanisms are also important to consider in the context of epilepsy. First, many types of epilepsies are associated with cognitive deficits, raising the possibility that these mechanisms are compromised in patients. Second, ictal and interictal activities may activate these mechanisms to trigger further network modifications. Third, these mechanisms may themselves be involved in the construction of epileptogenic neuronal networks.
Many processes can be evoked to alter information process transiently or permanently. Because information is coded in spike trains, it is essential to understand the mechanisms responsible for changing the output firing patterns in response to given synaptic inputs. The firing pattern of a neuron usually results from the interaction between synaptic inputs and ion (voltage-gated, Ca2+, ATP-gated, pH-gated, stretch-gated, etc.) channels. Any change at the synapse or ion channel level will result in a modified firing pattern. Changes in ion channels can modify firing patterns as efficiently as synaptic modifications do (in fact, both phenomena can occur simultaneously), be it during physiologic or pathologic conditions.9,39,108,114,117,137
Neurons use electrical and chemical processes to communicate at synapses. A chemical synapse is composed of four compartments (Fig. 1A): The presynaptic terminal, from which neurotransmitter is released; the postsynaptic terminal, which contains receptors for the neurotransmitter; the extracellular space between the pre- and postsynaptic elements; and glial cell processes, which contain various proteins, including transporters for the neurotransmitter. The arrival of an action potential in a presynaptic terminal usually results in the release of neurotransmitter that crosses the synaptic cleft to activate postsynaptic receptors. If these receptors are ionotropic, their activation leads to a flux of charged ions through the membrane. This flux of ions creates a current (I) that produces a change in membrane potential (V) (Fig. 1B). The term synaptic plasticity covers all of the mechanisms that ultimately result in a modification of V in response to an unconditioned stimulus (the activation of a presynaptic terminal by an action potential) following a conditioning stimulus. For example, at an excitatory glutamatergic synapse, postsynaptic depolarizing responses can be decreased on a short (short-term depression) or long (long-term depression, LTD) time scale, or increased (short-term facilitation or long-term potentiation, LTP).
Synaptic plasticity results from modifications that can take place at all four components of the synapse.
The presynaptic terminal, through changes in neurotransmitter release probability or the vesicular content of neurotransmitter; in this case, synaptic plasticity is said to have a presynaptic origin
The extracellular matrix (ECM), in which molecules can be involved in synaptic plasticity
The glial cells, which clear neurotransmitters (hence controlling the postsynaptic response) and which can release chemical messengers that can modify neurotrans-mission
The postsynaptic element, through morphologic alterations, changes in postsynaptic receptor density, or modifications of their intrinsic properties by intracellular processes (e.g., phosphorylation, interactions with partner proteins); in this case, synaptic plasticity is said to have a postsynaptic origin
This chapter focuses on the presynaptic terminal and the postsynaptic element.
Of note, multiple ways exist to induce synaptic plasticity and the underlying mechanisms are even more diverse.77 The goal of the chapter is not to provide an extensive review of the different mechanisms, but to provide representative examples. Synaptic plasticity mechanisms always depend on the conditioning stimulus (e.g., high- versus low-frequency stimulation), time (milliseconds to months following the conditioning stimulus), the developmental stage (e.g., mature versus immature neuronal networks), the brain region (e.g., synaptic plasticity at mossy fibers versus Schaffer collateral connections in the CA3 and CA1 hippocampal regions, respectively), and the type of neuron within a given region (e.g., γ-aminobutyric acid [GABA]ergic interneuron versus principal glutamatergic cells).
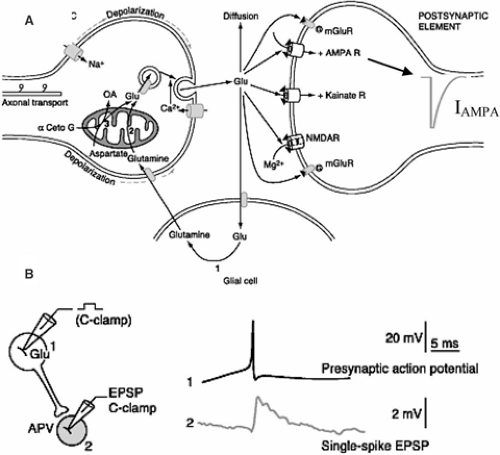 FIGURE 1. Synaptic transmission at a glutamatergic synapse. A: The four components of the synapse are the presynaptic and postsynaptic elements, the glial cell process, and the synaptic cleft. Depolarization of the presynaptic site (e.g., by an incoming action potential) results in Ca2+ influx that triggers fusion of a synaptic vesicle containing the neurotransmitter glutamate with the inner presynaptic membrane. Glutamate diffuses into the synaptic cleft to reach postsynaptic receptors (AMPA, NMDA, kainate, metabotropic). Activation of AMPA receptors triggers an influx of Na+ in the postsynaptic terminal, resulting in an inward current, IAMPA. Glutamate is pumped up by glial cells and presynaptic terminals by specific transporters. B: Example of a recording of a connected pair of neurons. The presynaptic glutamatergic neuron (1) is connected to neuron 2. When an action potential occurs in neuron 1, the membrane of neuron 2 is depolarized. This depolarization is due to an influx of Na+ through glutamate-activated AMPA receptors; that is, the transformation of IAMPA into a change in membrane potential, following Ohm’s law. Adapted from Hamon. |
Presynaptic Forms of Synaptic Plasticity
The first stage of information processing at the synapse takes place in the presynaptic terminal, from which neurotransmitter is released. Demonstrating that synaptic plasticity occurs at presynaptic terminals is technically challenging, because it requires direct recordings from the presynaptic terminal, an experimental technique that is still limited to very large presynaptic structures in the central nervous system (CNS).42 As a consequence, most presynaptic mechanisms are assessed indirectly, using recordings of the postsynaptic site. Despite this experimental constraint, presynaptic plasticity mechanisms have been established. Two forms of synaptic plasticity can be distinguished, short- and long-term.
Short-Term Plasticity
Neurons recorded in vivo display very different firing patterns in a brain state–dependent manner.105,115 In addition, firing patterns are also characterized by large intrinsic variability, even during stable brain state.105,115 The intrinsic properties of the presynaptic terminal determine how information is transmitted to the postsynaptic site. Here, the critical parameter is the interval between two action potentials—the interspike interval (Fig. 2). In response to closely spaced stimulations of a presynaptic terminal, some synapses display short-term facilitation. That is, the amplitude of the second response is larger than the first. Other synapses display short-term depression. That is, the amplitude of the second response is smaller than the first. Some synapses also display remarkable stability across a large range of interspike intervals, ensuring faithful neurotransmission (for example, the climbing fiber–Purkinje cell synapse in the cerebellum).13
Paired-Pulse Facilitation Mechanisms
Increased intracellular calcium (Ca2+) underlies paired-pulse facilitation.139 The favored mechanism proposes that the first action potential triggers a large and rapid rise in intracellular Ca2+, resulting in release of a neurotransmitter. This Ca2+ diffuses and equilibrates with the resting concentration of Ca2+ that was present before the action potential, thus giving rise to a residual Ca2+ concentration that will be cleared eventually. This residual Ca2+ facilitates neurotransmission when the second spike arrives at the presynaptic terminal (see Fig. 2).
Paired-Pulse Depression Mechanisms
The simplest model assumes a use-dependent exhaustion of the store of neurotransmitter-containing vesicles that are ready to be released in front of the postsynaptic site, but the issue remains controversial.139 Recovery from paired-pulse depression usually takes several seconds, but this can be facilitated by Ca2+ accumulation during high-frequency presynaptic activity.124 This phenomenon explains why neurotransmission does not fail totally during trains of action potentials at synapses displaying paired-pulse depression.
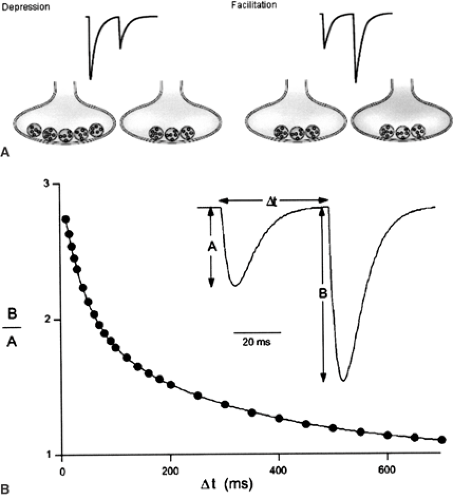 FIGURE 2. A: Paired-pulse depression and facilitation at glutamatergic synapses. A presynaptic terminal is stimulated to release glutamate, giving rise to an inward synaptic current of a given amplitude. Glutamate release is induced by a second, closely spaced, stimulation. If the amplitude of the second evoked current is smaller than the first one, the synapse is characterized by paired-pulse depression. A smaller number of vesicles ready to be released for the second stimulation may explain this phenomenon. If the second current is larger, the synapse is characterized by paired-pulse facilitation. An increased intracellular Ca2+ concentration (grey shading), thus increasing the probability of release in response to the second stimulation, may explain this phenomenon. Adapted with permission from Blitz DM, Foster KA, Regehr WG. Short-term synaptic plasticity: a comparison of two synapses. Nat Rev Neurosci. 2004;5:630–640. B: Simulation of paired-pulse facilitation showing that the amount of facilitation depends upon the interspike interval Δt. When the second stimulus occurs >600 ms after the first one, the amplitude of the second response is similar to the first. Facilitation is maximum at the shortest time intervals. Adapted with permission from Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. |
Modulation of Neurotransmitter Release by Presynaptic Receptors
The two short-term forms of plasticity involve mechanisms directly linked to the machinery for neurotransmitter release. The release of neurotransmitter can also be dynamically modified by chemical messengers acting via receptors located on the presynaptic terminal. For example, consider the mossy fiber terminals, the axons of hippocampal dentate granule cells that form giant synaptic boutons with CA3 pyramidal cells. Upon repetitive activation of mossy fiber terminals, released glutamate reaches high enough concentration to activate ionotropic (kainate; KA) and metabotropic (mGlu) receptors. The result of KA receptor activation is to increase excitability,111 thus facilitating transmitter release (i.e., directly participating in the strong paired-pulse facilitation seen at this synapse).26,112 Activation of type 2 mGlu receptors results in a decreased probability of glutamate release, probably via inhibition of presynaptic Ca2+ channels, thus limiting the
amount of facilitation.110 Other receptor types are also present on mossy fiber terminals, including adenosine A1 receptors. These receptors are tonically activated in vitro and result in a permanent low initial release probability of glutamate at this synapse.82 Synaptic vesicles can contain adenosine triphosphate (ATP) that, when released, is broken down into adenosine to activate presynaptic receptors.43 Whether A1 receptors act as autoreceptors (i.e., ATP is released by the terminal) or as heteroceptors (the source of ATP/adenosine is not the terminal136) is not known.
amount of facilitation.110 Other receptor types are also present on mossy fiber terminals, including adenosine A1 receptors. These receptors are tonically activated in vitro and result in a permanent low initial release probability of glutamate at this synapse.82 Synaptic vesicles can contain adenosine triphosphate (ATP) that, when released, is broken down into adenosine to activate presynaptic receptors.43 Whether A1 receptors act as autoreceptors (i.e., ATP is released by the terminal) or as heteroceptors (the source of ATP/adenosine is not the terminal136) is not known.
Use-dependent facilitation or inhibition of neurotransmission also involves other types of presynaptic autoreceptors, including N-methyl-D-aspartic acid (NMDA) receptors51 and α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors109 at glutamatergic terminals, and metabotropic GABAB receptors at GABAergic terminals.29 Facilitation may be the result of an increase in internal Ca2+ concentration resulting either from direct Ca2+ entry through Ca2+-permeable receptors (e.g., NMDA) or Ca2+ voltage-gated channels opened by the receptor activation–induced membrane depolarization. Inhibition may result from downregulation of Ca2+ voltage-gated channels by G-proteins following activation of metabotropic receptors. To make things even more complex, presynaptic terminals can possess heteroreceptors that are activated by chemical messengers not released by the terminal itself. For example, repetitive activation of glutamatergic parallel fibers in the cerebellum activates GABAergic interneurons. GABA is released in large enough quantities to activate presynaptic GABAB receptors located on parallel fiber terminals contacting Purkinje cells, thus decreasing the probability of glutamate release by a Ca2+-dependent mechanism.31
Conclusion on Short-Term Plasticity Phenomena
Presynaptic terminals are complex information-processing units. They play a crucial role in shaping postsynaptic responses by increasing or decreasing neurotransmitter release as a function of the timing of the spike train reaching the presynaptic terminal. They use multiple mechanisms to modify their output depending on the type of activity they receive, the molecular properties of their release machinery, the biophysics of their ionic channels, and the type of autoreceptors they express. Through activation of heteroreceptors, presynaptic terminals also sense the activity of neighboring terminals, which can be of a different type. The properties of a terminal depend on its origin, target, and developmental stage.139 These properties play a very important role in information processing, for example, by allowing proper routing of information in the network by adequately shaping information transfer.104 Any alteration in the mechanisms that control paired-pulse facilitation or depression at a given synapse will result in a dramatic change in function. Such possibilities should be considered when investigating neurotransmission and information processing in pathologic conditions.
Long-Term Plasticity
Among the many loci at which a presynaptic form of synaptic plasticity has been found, LTP at the mossy fiber has received considerable attention.91 High-frequency stimulation (e.g., 100 pulses at 100 Hz) of mossy fibers leads to a permanent enhancement of the postsynaptic response. Three questions are in order: Where is LTP expressed: Pre- or postsynaptically? What
are the expression mechanisms (what makes the response larger after the conditioning stimulus)? and, How is this phenomenon induced by the conditioning stimulus?
are the expression mechanisms (what makes the response larger after the conditioning stimulus)? and, How is this phenomenon induced by the conditioning stimulus?
Presynaptic Expression of Long-Term Potentiation
The expression of LTP at mossy fiber cell synapses is due to an increase in neurotransmitter release. As mentioned previously, presynaptic mechanisms can only be inferred indirectly, and various approaches have been used to support a presynaptic rather than postsynaptic locus. The arguments supporting the former are a decrease in paired-pulse facilitation,132,135 a decreased failure rate,73,132 and a change of the coefficient of variation (CV)73,132 following LTP induction, all classical properties expected to occur if the probability of neurotransmitter release is increased. The failure rate is directly linked to release probability. Transmitter release is a probabilistic phenomenon, because an action potential does not always trigger vesicle fusion. Some synapses (such as the cerebellar climbing fiber) are reliable, having a close to 1.0 probability, whereas others have a low release probability (such as the mossy fiber, in part due to the tonic activation of A1 receptors, as described earlier). Repetitive activation of presynaptic terminals with <1.0 release probability gives rise to failures—no postsynaptic response is recorded despite the presence of an action potential in the presynaptic terminal. A decreased number of failures indicates an increased release probability. The CV is the mean postsynaptic response divided by the standard deviation of the response (using repetitive stimulation). This parameter also depends strongly on release probability. Other arguments in favor of a presynaptic mechanism have been proposed, including the measurement of postsynaptic NMDA responses,126 and the monitoring of extracellular glutamate concentration directly76 or indirectly using glial cells.58
Expression Mechanisms
The mechanisms that underlie increased neurotransmitter release at mossy fiber synapses are not fully understood and still the subject of debate.20 A simple scheme has been proposed91 in which the conditioning stimulus leads to increased intracellular Ca2+ in the presynaptic terminal,119 activation of α1E-subunit–containing R-type Ca2+ channels.17 The rise in Ca2+ would activate Ca2+/calmodulin-activated adenylyl cyclase (AC)-1, which is highly expressed in mossy fiber terminals.131 Activation of AC1 would result in a rise in cyclic adenosine monophosphate (cAMP), leading to the activation of protein kinase A (PKA). Interfering with any one of these steps alters mossy fiber LTP.50,73,119,123,125 How this leads to enhanced neurotransmitter release, and how LTP is maintained over the long-term, remain to be established.
Induction Mechanisms
In contrast to postsynaptic expression of LTP at Schaffer collateral synapses (see the next section), the induction of mossy fiber LTP does not require activation of postsynaptic NMDA receptors.45 The key player is Ca2+, because its removal or strong buffering prevents LTP.119 The next issue is to determine if the induction depends on a rise of Ca2+ in the presynaptic terminal, the postsynaptic unit, or both. Several studies have reported that a rise in postsynaptic Ca2+ is necessary,128,133 whereas others have shown the opposite.81,135 This issue is functionally important, because a postsynaptic induction mechanism implies that a retrograde messenger must be sent by the postsynaptic cell to alter synaptic release in the presynaptic terminal. A reasonable way to explain this discrepancy (and others) is that different induction mechanisms exist, and their activation depends on the experimental conditions present. The role of KA and mGlu receptors is also fiercely debated, with contradictory results regarding their direct involvement or modulatory role.14,16,81,133
Long-Term Depression at Mossy Fiber Synapses
Synaptic responses are enhanced at mossy fibers following tetanic stimulation, and they are decreased (i.e., LTD) following low-frequency stimulation (e.g., 15 minutes at 1 Hz).61,122 The expression of LTD is also presynaptic, and seems to involve mirror mechanisms when compared to those described for LTP at the same synapse—a decrease in adenylyl cyclase and PKA activity.122 Using mechanisms similar to those involved in LTD, after LTP, it is possible to reverse the synaptic responses to those of preconditioning stimulus values, a phenomenon known as depotentiation.48
Target Specificity of Synaptic Plasticity
The forms of synaptic plasticity described so far concern the synapse between mossy fibers and CA3 pyramidal cells. However, mossy fibers produce small filopodial extensions that contact CA3 GABAergic interneurons.2 Indeed, GABAergic interneurons are the main target of mossy fibers, because 10 times more filopodial extensions than mossy fiber boutons contact pyramidal cells.2 High-frequency stimulation induces LTD at mossy fiber–interneuron synapses, which express postsynaptic Ca2+-permeable AMPA receptors.67 As in mossy fiber–pyramidal cell LTP, this form of LTD is NMDA receptor–independent, depends on type 7 mGlu receptor and protein kinase C (PKC), and is expressed presynaptically.68,98 However, it requires a rise in postsynaptic Ca2+, which means the involvement of a retrograde messenger that has yet to be identified. This illustrates that the same experimental protocol can trigger opposite synaptic modifications onto two different targets.
To illustrate further the importance of target specificity, it is interesting to note that the same conditioning stimulus triggers a different form of LTD at those mossy fiber–interneuron synapses that express postsynaptic Ca2+-impermeable AMPA receptors.67 This form of LTD requires a postsynaptic rise in intracellular Ca2+ via the activation of an NR2B-subunit lacking NMDA receptors.67 The expression involves a downregulation of postsynaptic AMPA receptors.68 (Postsynaptically expressed long-term plasticity is discussed later.)
Other Examples of Plasticity Expressed Presynaptically
LTP can be expressed presynaptically at other glutamatergic synapses (e.g., in the cerebellum at the parallel fiber–Purkinje cell synapse44 or at corticothalamic synapses19). Mechanisms are similar to those described previously; and require involvement of adenylyl cyclase, cAMP, and PKA.19,44
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


