The Dandy-Walker Complex and Arachnoid Cysts
13.1 Dandy-Walker Complex
The Dandy-Walker complex is a heterogeneous group of anatomically and possibly embryologically related disorders that have in common a distortion of midline cerebellar anatomy and a large posterior fossa cyst, usually in communication with the fourth ventricle. The complex often appears in conjunction with other central nervous system (CNS) abnormalities, especially agenesis of the corpus callosum. Pathologically, the enlarged posterior fossa fluid collection can expand, compressing other posterior fossa structures and producing hydrocephalus. Surgical management is usually directed at treating the hydrocephalus or the posterior fossa cyst, or both, either by cerebrospinal fluid (CSF) shunt diversion or by endoscopic techniques.
13.1.1 Anatomy and Pathogenesis
Anatomy
Three described and likely related entities, (1) Dandy-Walker cyst (DWM), (2) Dandy-Walker variant (DWV), and (3) persistent Blake pouch, have been lumped together as Dandy-Walker complex (DWC). A fourth diagnosis, mega cisterna magna, may belong, as well1–4 (▶ Fig. 13.1). In addition, midline posterior fossa arachnoid cysts are frequently part of the differential diagnosis of posterior fossa cystic lesions.
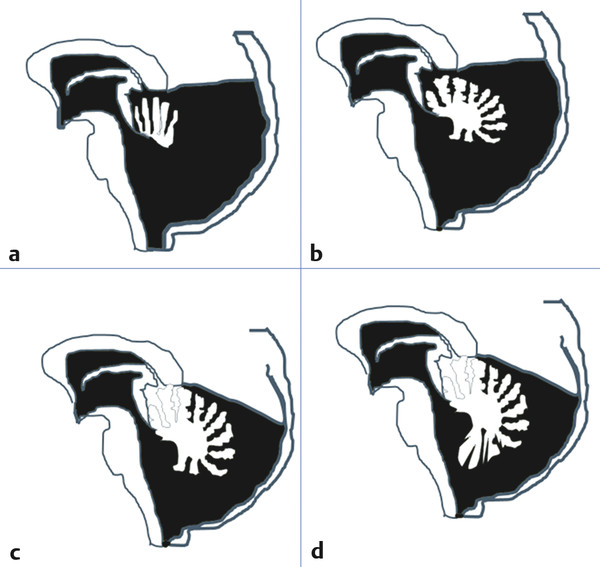
Fig. 13.1 The continuum of the Dandy-Walker complex. (a) Dandy-Walker malformation: posterior fossa enlarged, with minimal to no vermian remnant present and rotated superoanteriorly. Note: The corpus callosum is shown in this diagram for ease of understanding. However, agenesis of this structure frequently coexists with the Dandy-Walker malformation. (b) Dandy-Walker variant: posterior fossa enlarged, superior cerebellar vermis partially preserved and lobulated. (c) Persistent Blake pouch: posterior fossa volume only modestly enlarged, cerebellar vermis fully present and lobulated, inferior vermis compressed, vallecula enlarged. (d) Mega cisterna magna: posterior fossa volume modestly enlarged or normal, cerebellar vermis fully present and lobulated, vallecula normal in size.
(Modified from Yildiz H, Yazici Z, Hakyemez B, Erdogan C, Parlak M., Evaluation of CSF flow patterns of posterior fossa cystic malformations using CSF flow MR imaging. Neuroradiology 2006;48(9):595–605.)
Anatomically, DWM consists of a large cystic fluid collection that appears to arise out of an expanded fourth ventricle with an associated lack of the cerebellar vermis. The posterior fossa is typically enlarged, with elevation of the tentorium and the torcular herophili and associated confluence of the venous sinuses. The cerebellar hemispheres are present, although they may be compressed by the cyst, which may also compress the brainstem. The aqueduct of Sylvius is variably obstructed. Hydrocephalus is present in many, although not all, patients and may be present with or without an obstructed aqueduct.1,4 Radiographically, initially based on axial computed tomography (CT), DWM came to be defined as an entity in which no apparent vermis was present. DWV has been defined as the presence of some residual superior vermis in the setting of a midline posterior fossa fluid collection that appears to be in communication with the fourth ventricle through a significantly enlarged vallecula, usually with a lesser degree of expansion of the posterior fossa.4,5 In DWV, the cerebellar vermian remnant is rotated superoanteriorly. CSF communication between the expanded fourth ventricle and the subarachnoid space is variably present, and hydrocephalus is less common.6 A persistent Blake pouch has been more recently defined as a posterior fossa cyst in communication with the fourth ventricle, often through a mildly expanded vallecula. The cyst walls are sometimes visible on sagittal magnetic resonance (MR) imaging as distinct from the subarachnoid space. The inferior vermis is elevated and compressed to a degree, but its anatomical divisions as seen on a sagittal plane MRI are preserved.2,3 Lastly, a mega cisterna magna appears as an expansion of the subarachnoid spaces of the posterior fossa. It is distinguished anatomically from a posterior fossa arachnoid cyst by its free communication with the rest of the subarachnoid space and the fourth ventricle. The cerebellum appears normal or anteriorly displaced, and the vallecula is only minimally or not enlarged. The inferior vermis is not focally compressed as it appears to be in a persistent Blake pouch.6 The reader should be aware that considerable disagreement exists regarding the precision of these definitions.7
Pathogenesis
The combination of cerebellar dysgenesis, a large posterior fossa cyst, and hydrocephalus was first described by Sutton in 1887.8 Walter Dandy and Kenneth Blackfan reported a similar case in their 1914 treatise on CSF physiology.9 They observed that the foramina of Luschka and Magendie appeared to be absent. Dandy and others reported case series in which the etiology of the cystic dilatation in the posterior fossa was identified as occlusion of the outflow foramina of the fourth ventricle.10,11 However, other authors identified similar cases in which the foramina of Luschka and Magendie were patent.12 As an alternative to the hypothesis of foraminal atresia, Benda proposed that maldevelopment of the cerebellar vermis was likely responsible.13 Supporting this, Brodal and Hauglie-Hanssen, in their seminal paper, presented two autopsy cases with detailed dissections.14 The posterior vermis was underdeveloped in both, while the more anterosuperior vermian structures were preserved. The posterior vermian remnant was noted to be continuous with the cyst lining, which had some ependymal features and which they felt was continuous with the rest of the fourth ventricular ependyma. They noted that in the human embryo, the cerebellar anlagen fuse and the vermian divisions form well before the fourth ventricular outflow foramina open. The fusion begins superiorly and proceeds inferiorly. Given the maldevelopment of the vermis seen, they argued that the primary defect must be with the precursor of this structure, the anterior membranous area (AMA).14 The appeal of this hypothesis is that it appears consistent with the variability of vermian dysgenesis seen across the DWC.
In the developing embryo, the posterior membranous area (PMA) is separated from the AMA by the developing choroid plexus. The PMA normally regresses, with the foramen of Magendie developing in its place. Tortoni-Donati et al, and later Calabrò et al, postulated that the Blake pouch, a small expansion of the PMA seen in utero, can persist and expand rather than regress to give rise to a posterior fossa fluid collection in communication with the fourth ventricle in which the inferior vermis is appropriately lobulated, if compressed.2,3,15 It has been further argued that the mega cisterna magna also could be categorized as a PMA developmental abnormality in which the developing PMA cyst eventually becomes connected with the subarachnoid space.3,15
The reader should be aware that considerable controversy exists about whether the radiographic and clinical features occurring in DWM, DWV, and persistent Blake pouch should be considered discrete or continuous. Barkovich et al, noting the variability in radiographic findings, have argued that these entities should be viewed as a continuum.1,4 Geneticists and others focused on the various associated malformations seen in various aspects of DWC have argued for a more compartmentalized view.16
13.1.2 Epidemiology
In a population-based study of members of the Saudi Arabian Armed Forces and their families, DWM had an incidence of 1 in 100,000 live births.17 Long et al reported another population-based study, from northern England, wherein DWMs and DWVs were identified in utero in 1 per 11,574 live births (approximately 9 per 100,000). Most were identified on prenatal ultrasound, and 47% had associated congenital malformations. Prenatal termination, fetal demise, or neonatal demise occurred in the 43 of 47 prenatally diagnosed cases.18 Hirsch et al derived an incidence of 3 to 4 per 100,000 live births, calculating this from the incidence of hydrocephalus and the frequency of DWMs in their cohort.19 Most, although not all, studies show an increased incidence in females.18–21 Familial cases of DWM have been reported, but without clear inheritance patterns.22–24
Reefhuis et al, using the National Birth Defects Prevention Study, noted the use of clomiphene citrate to be associated with an increased likelihood of DWMs (adjusted odds ratio [OR], 95%; confidence interval [CI], 4.44, 1.7–11.6) among a variety of other malformations, including congenital heart disease, craniosynostosis, cloacal exstrophy, and omphalocele.25 Prenatal exposures to a number of other agents, including benzodiazepines and coumadin, have been reported in association with DWMs, but without sufficient power to draw conclusions.
A wide variety of genetic abnormalities have been reported in patients with DWM. Chromosome 3, specifically 3q24, which includes two genes encoding for cerebellar formation, has been implicated. Deletions of this region in animal models produce an appearance similar to DWM. Trisomy of chromosomes 9, 13, and 18 has been reported frequently in the genetic analysis of affected individuals.26
13.1.3 Clinical Presentation
In neurosurgical series, the most common presentation is hydrocephalus, with rates of up to 80% of patients with DWM.19,20,27,28 In a population of children all with DWV, Sasaki-Adams et al reported a 29% rate of ventriculomegaly, but only 14% of the patients required surgical treatment.21 The incidence in nonsurgical series is likely lower. Most children present within the first year or two after birth. In many, the diagnosis is known from prenatal studies. The specifics of the presentation depend on the patient’s age; infants present with progressive macrocrania, bulging fontanels, and upward gaze palsy. The speed at which patients become symptomatic is not uniform. In the series of Raimondi et al, children presented between 2 and 9 months of age.29 Hirsch et al reported that 80% presented before 1 year of age, but 7 of 40 required their first treatment after 2 years of age.19 Additional symptoms may include bilateral nystagmus, alterations in speech cadence, weakness, and respiratory difficulties.29 Opisthotonos has been reported, presumedly due to brainstem compression.30 Older patients who survive infancy and can be assessed in more detail often show a striking lack of ataxia relative to the degree of cerebellar malformation. Adult presentation has been reported with both hydrocephalus and cerebellar symptoms.31,32 As noted above, the majority of children born with DWMs will have associated abnormalities, which may dictate their clinical presentations. Some patients are asymptomtic and identified incidentally.33
13.1.4 Associated Conditions
Both CNS and systemic anomalies occur in association with DWC. Depending on the setting of the study and the specifics of the malformation studied, between 50 and 80% of patients are affected. When component diagnoses of DWC are compared, malformations are very common in both DWM and DWV, and it is not yet clear whether DWV has a much lower incidence of such associated malformations, as is sometimes supposed. Has et al reported a prenatal series of 64 fetuses with DWM and 14 with DWV. Because of the setting, 45% of the parents were consanguineous. Excluding hydrocephalus, 23% (DWM) and 42% (DWV) had CNS malformations, most commonly agenesis of the corpus callosum in 10%. Non-CNS abnormalities were present in 44% and 64% of the patients with DWM and DWV, respectively.34 Sasaki-Adams reported on a group of 24 children with DWV followed over a mean 5 years, with four requiring shunt placement for hydrocephalus. Associated CNS abnormalities included agenesis of the corpus callosum (21%), schizencephaly, seizures, and cortical blindness. Non-CNS malformations were most commonly cardiac (42%) and gastrointestinal (33%).21 When diagnoses were made in utero, 56% of the patients with mega cisterna magna in the aforementioned population study from northern England were found to have associated anomalies, although their developmental outcome was dramatically better (25 of 29 were normal) than that for patients in this study with DWM or DWV. Given the midline nature of DWC, it is not surprising that many of the associated abnormalities are also located in the midline, including occipital encephaloceles.19 The box “Syndromes Associated with the Dandy-Walker Complex” lists a number of other neurologic abnormalities that have been noted.
Syndromes Associated with the Dandy-Walker Complex
Aase-Smith syndrome
Beemer-Langer syndrome
Chondrodystrophia calcificans congenita
Chronic hereditary polyneuropathy
Coffin-Siris syndrome
Cornelia de Lange syndrome
DiGeorge syndrome
Down syndrome
Ellis-van Creveld syndrome
Facioauriculovertebral syndrome
Fetal akinesia deformation sequence
Genoa syndrome
Goldston syndrome
Juberg-Hayward syndrome
Kallmann syndrome
Klippel-Feil syndrome
Marden-Walker syndrome
Meckel-Gruber syndrome
Neurofibromatosis
Neurocutaneous melanosis
Oculocerebrocutaneous syndrome
Opitz C syndrome
Oral–facial–digital syndromes
Partial trisomy 22
PHACES (posterior fossa malformations, hemangioma, arterial anomalies, cardiac defects, eye anomalies, sternal defects) syndrome
Renal–hepatic–pancreatic dysplasia
Rokitansky syndrome
Ritscher-Schinzel (3C: cranial vault, cerebellar, cardiac) syndrome
Sjögren-Larsson syndrome
Smith-Lemli-Opitz syndrome
Tetrasomy 9
Trisomy 9
Turner syndrome
Walker-Warburg syndrome
Yunis-Varon syndrome
DWC has been seen in a wide variety of named syndromes, which are listed above. Most of these appear in isolated case reports, but several appear with more regularity, including PHACES syndrome (posterior fossa abnormalities, hemangioma of the face, arterial abnormalities, coarctation of the aorta, eye abnormalities, sternal defects)35,36 Ritscher-Schinzel (3C) syndrome (cranial vault, cerebellar, cardiac)37 neurocutaneous melanosis38 and Meckel-Gruber syndrome (renal cystic dysplasia, CNS malformations, polydactyly, hepatic defects, pulmonary hypoplasia). In the latter, a Dandy-Walker cyst can replace the more common occipital encephalocele.39,40 Source: From Raimondi AJ, Sato K, Shimoki T. The Dandy-Walker Syndrome. Basel, Switzerland: S Karger; 1984:21–45, as presented in Wilkinson C, Winston K. Congenital arachnoid cysts and the Dandy-Walker complex. In: Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. 2nd ed. New York, NY: Thieme Medical Publishers; 2007:165.184
| Middle, anterior cranial fossa | Porencephalic cyst Epidermoid cyst Loculate subdural hygroma Schizencephaly Neurocysticercosis Cystic neoplasm Choroid fissure cyst Vascular lesion/aneurysm |
| Sellar and suprasellar | Craniopharyngioma, other cystic tumors, Rathke cleft cyst Dermoid cyst |
| Midline posterior fossa | Dandy-Walker complex (malformation/variant/persistent Blake pouch) Mega cisterna magna Neurenteric cyst |
| Quadrigeminal plate region | Pineal cyst Tumor cyst Neuroepithelial cyst |
| Cerebellopontine angle | Epidermoid cyst Cystic cerebellopontine angle tumor Infectious cyst (should be intra-axial) |
| Intraventricular | Choroid plexus cyst Ependymal cyst Colloid cyst Neurocysticercosis |
| Source: Modified from Osborn AG, Preece MT. Intracranial cysts: radiologic-pathologic correlation and imaging approach. Radiology 2006;239(3):650–664.185 | |
13.1.5 Radiology and Treatment Planning
Radiographic findings in DWC mirror the anatomical details already noted. The enlarged posterior fossa and thinned cranium can be seen on plain skull radiographs. In the normal individual, the torcular herophili lies below the lambda on skull X-rays. The vascular grooves created by the sinuses are seen below the lambdoid sutures. Lambdoid–torcular inversion occurs with the expansion of the posterior fossa seen in DWC, such that the confluence of the sinuses can lie above the lambda and is seen above the lambdoid sutures on plain X-ray. Angiographically, in the arterial phase, the posterior inferior cerebellar arteries may be absent.41 In the venous phase, the oblique course of the transverse sinuses and upward course of the straight sinus are noted.29
The accuracy of prenatal ultrasound for the diagnosis of DWM/DWV has ranged in the literature from as low as 40% to up to 92%.18,42 Diagnosis before 18 weeks of age may have a higher error rate.18 Fetal MR imaging is increasingly being performed. Although MR imaging has significant advantages postnatally, the accuracy of prenatal diagnosis in early gestation has been questioned. Patek et al reported on 59 fetuses found on MR imaging to have posterior fossa abnormalities demonstrating the spectrum of findings seen in DWC. When the patients were assessed on postnatal imaging, for those with findings equivalent to DWV, the sensitivity was 50% (5 of 10) when the study was performed before 24 weeks, compared with 100% (9 of 9) when MR imaging was performed after 24 weeks.43 Postnatally, however, MR imaging is clearly the diagnostic study of choice, revealing the posterior fossa anatomy considerably better than ultrasonography, and MR imaging is more sensitive to other CNS abnormalities seen, such as those listed in the box “Syndromes Associated with the Dandy-Walker Complex.”
When the treatment of patients with DWC is being planned, knowing which of the various fluid spaces are in communication may be helpful. The aqueduct of Sylvius may appear obviously occluded on standard MR images. However, CT after contrast injection into the ventricular system or cyst may be a more definitive study, although at the cost of radiation exposure, usually in a young patient. As an alternative, phase-contrast MR imaging has been proposed to determine the relative patency of the cystic structures in DWC. Yildiz et al reported a very high correlation rate between phase-contrast cine MR imaging and CT cisternography in delineating the anatomy of the DWC variations and in differentiating between these and posterior fossa arachnoid cysts.6
13.1.6 Treatment
DWC spans a variety of clinical presentations and therefore treatment needs. DWM is associated with hydrocephalus in 80% of cases, so most patients with DWM become symptomatic and require treatment at some point in early childhood. Surgical options include CSF shunt placement, endoscopic management, and open surgical treatment. Because of the high morbidity associated with early open surgical treatments, CSF shunt placement became the principal treatment as soon as shunts were readily available.29,44
The debate about whether a CSF shunt should be placed into the ventricular system, the cyst, or both has been continuing since the onset of shunt treatment and remains unresolved, save for agreement that placing the catheter into one of the spaces necessitates either preoperative demonstration of communication between the spaces or careful postoperative observation for evidence of loculation formation and noncommunication. Supporting treatment of just one compartment, Mohanty et al noted that 23 of 26 patients with DWM in their series had a patent aqueduct on MR imaging.45 Hirsch et al reported success with isolated shunt systems in either compartment.19 However, Asai et al, reporting the Toronto Hospital for Sick Children series of 35 patients, noted that 9 of 21 patients (43%) treated with isolated ventricular shunts developed noncommunication with the cyst, necessitating cyst shunt treatment.46 Osenbach and Menezes reported that 75% of patients with DWM had noncommunication requiring shunting of both compartments.28 Naidich et al identified radiographic findings of downward herniation of the supratentorial contents after cyst decompression or lateral ventricular shunt obstruction in 25 patients with DWM.47 Kumar et al reported that 9 of 28 patients (32%) treated with ventricular shunts required subsequent cyst shunts, compared with 6 of 7 patients (86%) with a cyst shunt who later required ventricular shunts.27 Mohanty et al reported that 12 of 24 cyst shunts failed, compared with 4 of 21 ventricular shunts. Five patients went on to require shunting of both compartments, and endoscopic techniques were also used.45 Those advocating shunt placement in both compartments have generally argued for a two-limbed shunt so that both the posterior fossa and supratentorial space are exposed to the same intracranial pressure (ICP).29
A number of surgical pitfalls must be avoided during CSF shunt placement in this population. If a cyst catheter is to be placed, a lack of brain parenchyma to pass the catheter through increases the incidence of postoperative CSF leak or subcutaneous collection, with this occurring in 29% of patients with cyst shunts in one case series.45 A very small durotomy for catheter placement, appropriately compressive dressings, and potentially an adjustable-pressure valve with a low initial setting may help limit the potential for this complication. The enlarged posterior fossa in DWM means that the standard external landmarks for posterior ventricular shunt placement are unreliable. In addition, the dural venous anatomy is distorted, and there is risk that a bur hole intended for a supratentorial catheter may be placed over the transverse sinus. Intraoperative ultrasound can be a valuable adjunct in patients with open fontanels to guide catheter placement. Simultaneous single-catheter placement into both the ventricular and cystic cavities has been reported with ultrasound guidance.48 More recently, the neuroendoscopically assisted cannulation of ventricular and cyst cavities with a single catheter has been advocated.49,50
Shunt failure is reported in all surgical series. In the above-cited case series, the shunt failure rates range from 4 to 60% over variable follow-up. If one compares these rates with those in similarly structured case series for other types of hydrocephalus, the results are not clearly different. Other complications include brainstem or cranial nerve injury from catheter placement, intracystic hemorrhage, and catheter migration into or out of the posterior fossa cyst.51,52 Chronic traction on the brainstem from a cyst catheter adherent to it, with neurologic injury, has been reported.53
Endoscopic third ventriculostomy (ETV) in patients with DWM has been reported as part of larger series.45,54,55 Given that some patients have patent aqueducts, and assuming obstruction at the outflow of the fourth ventricle or cyst, this should be a reasonable choice. Distortion of the third ventricle and a narrow prepontine subarachnoid space increase the complexity of such procedures, but success has been reported in what would otherwise appear to be challenging cases.56 In cases of noncommunication between the ventricles and DWM, endoscopic cyst fenestration to the ventricular system has been advocated as an addition to ETV. Mohanty et al used endoscopic techniques in addition to CSF shunt placement in 21 patients, with 5 failures (23%) requiring alternative procedures. The authors describe ETV, cyst fenestration, and guided stent placement through the superior vermian remnant from the supratentorial space.45,57 Warf et al reported on their experience in Uganda treating DWM/DWV with ETV and choroid plexus coagulation in 45 children who had a mean age of 5 months at treatment. With a minimum of 6 months’ follow-up (mean, 24 months), 74% of the patients had not required an additional procedure. The authors observed that 39 of 41 patients had an open aqueduct as seen endoscopically at surgery.58
Direct surgical resection of the cyst wall was the initial procedure performed before the availability of CSF shunts. It had a high failure rate and a high associated mortality rate. Summarizing nine case series reported between 1947 and 1984, Hirsch et al noted a 75% failure rate and 10% surgical mortality rate.19 However, a number of more recent publications detail, albeit in small numbers of patients, less adverse outcomes with open techniques than those in older reports.59,60
13.1.7 Outcome
Survival
The analysis of outcomes for patients with DWC is complicated by the variability of the complex itself and the associated malformations. In addition, selection and survivor biases are clearly complicating factors in addressing this question. Using the Congenital Malformations Registry of the New York State Department of Health, Salihu et al reported a 25% mortality rate by the end of the first year of life in 196 cases. The death rates were 16% for patients with isolated DWS, 32% for those with DWS and one additional organ system anomaly, and 42% for those with involvement of two or more additional organ systems.61
Intellectual Outcome
Neurodevelopmental outcomes are similarly variable. Among long-term survivors from before 1984, Hirsch et al reported that 49% had IQ scores above 90.19 A number of authors report a relationship between the degree of cerebellar dysgenesis and intellectual outcome. Gerszten and Albright reported that 45% of 120 patients with DWS had normal intelligence. Using CT images to measure the ratio of cerebellar volume to posterior fossa volume, they noted no relationship between the ratio and intellectual outcome.62 However, using MR imaging, Boddaert et al reported that in 21 children with DWC, those with normal cerebellar lobulation had an 82% rate of normal intelligence, whereas no child with abnormal cerebellar lobulation had normal intelligence.63 Klein et al also reported a much more favorable intellectual outcome with only partial as opposed to severe vermian abnormalities.64
With specific reference to DWV, developmental outcomes have generally been assumed to be better than those in DWM, but as in DWM, a wide range of outcomes is reported, and associated congenital anomalies appear to play a major role. Sasaki-Adams noted that 5 of 6 patients with isolated DWV had normal development, but 10 of 15 with DWV and associated abnormalities were impaired, 6 of them severely.21 Mega cisterna magna is generally thought of as a benign condition, and neurodevelopmental assessments in such patients support this view, with the exception that, as in other DWC entities, associated abnormalities play a significant role.43,65
13.2 Arachnoid Cysts
Intracranial arachnoid cysts are arachnoid-lined expansions of the normal subarachnoid and, in some cases, intraventricular spaces. They are more common in boys than in girls, more often left-sided than right-sided, and more often above the tentorium than below it. Common sites include the middle cranial fossa, the suprasellar region, the quadrigeminal plate region, and the posterior fossa. Arachnoid cysts are usually asymptomatic, but they occasionally cause symptoms and require treatment. Treatment options include craniotomy for microsurgical cyst fenestration, endoscopic fenestration, and CSF shunt placement. The indications for treating an arachnoid cyst must be carefully considered in light of the natural history of these lesions.
13.2.1 Pathogenesis
The first clear description of an intracranial arachnoid cyst is usually credited to Richard Bright in Reports of Medical Cases, published in 1831. Bright identified the intra-arachnoidal location of the fluid collection and the variable size of cysts found in different individuals. He also noted the remodeling of the brain and bone caused by the cyst. He identified most of these cysts as likely chronic, with little propensity for change.66,67 A variety of explanations for the origin of the arachnoid cyst have subsequently been proposed, including posttraumatic and infectious etiologies.68–70 Most cysts, however, likely have a congenital origin, particularly those diagnosed in childhood. Microscopically, the cyst appears as a duplicated layer of otherwise normal arachnoidal tissue that forms the cyst wall.68,71 In their ultrastructural study, Rengachary and Watanabe identified a split in the arachnoid membrane at the margin of the cyst, associated thickening of the collagen in the arachnoid cyst wall, hyperplasia of arachnoid cells in the cyst, and the absence of the typical spiderlike processes that define the normal arachnoid membrane. They postulated that the cysts develop in utero.72 The exact timing of cyst development in utero is uncertain. Studies of associated developmental venous anatomy suggest that cysts in the sylvian fissure may form in the middle of the first trimester.73 Prenatal imaging has similarly demonstrated arachnoid cysts as early as the first trimester.74 Bannister et al reported on 15 fetuses in a single-institution review. Five were diagnosed before 20 weeks. Thirteen cysts were supratentorial.75 The authors noted that ultrasound diagnosis was sometimes difficult, with two of four aborted fetuses in this series having other etiologies accounting for the ultrasound findings. Arachnoid cysts have also been evaluated with fetal MR imaging.76–78 Case series of children with prenatally diagnosed arachnoid cysts report a higher incidence of associated abnormalities and the need for surgical treatment than do series of children with a postnatal diagnosis.75,76
From a developmental standpoint, the predominance of a middle fossa location, the left-sided predominance, and the male predominance seen in intracranial arachnoid cysts lack a proven explanation. Wester conjectured that during the anterior growth of the temporal lobe adjacent to the frontal tissues, which creates the sylvian fissure, the leptomeningeal tissues are carried along with the cerebral lobes, so that the temporal and frontal arachnoid tissues are placed in apposition. If the developing frontal and temporal arachnoid tissues fail to fuse, cyst formation can then occur.79 However, cysts in other locations cannot be readily explained by a similar mechanism. No clear hypothesis for the male predominance or left-sided predominance has been advanced.
Fox and Al-Mefty proposed that suprasellar arachnoid cysts form from an upward diverticulum of the membrane of Liliequist.80 Open or endoscopic surgical treatment of these lesions frequently discloses a much more thickened and tough membrane than is seen in middle fossa arachnoid cysts. Rests of arachnoidal tissues are periodically present in the ventricular system and are thought to account for the rare intraventricular meningioma. These cells are presumably also the source of the equally rare intraventricular arachnoid cyst.
Three theories have been advanced for how fluid accumulates in an arachnoid cyst. First, based on surgical observation, arachnoid tissues have been seen to form a one-way valve, admitting fluid during one phase of the cardiac cycle but not allowing it to subsequently escape. Second, cyst fluid may accumulate as the result of an osmotic gradient. Third, the arachnoid membranes may actively secrete the cyst contents. Schroeder and Gaab provide endoscopically acquired photographs of a slit valve in the base of a suprasellar arachnoid cyst.81,82 Similar findings have been noted on cine MR imaging.83 The contents of an arachnoid cyst are typically thought of as being identical to spinal fluid. However, increased cyst protein concentrations were noted in a group of 54 pediatric patients undergoing open surgical treatment.84 The authors argued that this supported an osmotic mechanism. However, the patients did not act as their own comparators for the study, with reference CSF values being used instead. Berle et al, conversely, provided an analysis of fluid drawn from both the subarachnoid space and the arachnoid cyst in 15 patients undergoing surgical treatment. Although most electrolyte concentrations were similar, increased concentrations of phosphate, along with decreased concentrations of protein, ferritin, and lactate dehydrogenase, were noted in the cyst fluid. The authors argued that a ball-valve mechanism for filling would not allow these differences. Fluid osmolarity was similar, arguing against an osmotic force. The authors interpreted these data as supporting fluid secretion by the cyst walls or some other active transport mechanism.85–87 Earlier studies had also suggested an active transport mechanism in cyst fluid formation.88 It is of course possible that several mechanisms may be in operation.
Trapped fluid within the arachnoid cyst displaces and distorts the adjacent neural and neurovascular structures. The degree to which neural structures tolerate the sometimes significant displacement is remarkable.89 Even very large cysts with significant brain displacement are regularly found incidentally, particularly in the middle and anterior cranial fossae. Conversely, large midline cysts, particularly in the suprasellar region, frequently produce endocrine dysfunction and hydrocephalus.90–92 Quadrigeminal plate arachnoid cysts are frequently associated with hydrocephalus. A variety of functional studies have addressed whether large supratentorial arachnoid cysts distort normal cerebral metabolism, with conflicting results. Wester and Hugdahl reported changes in measures of verbal laterality and handedness following surgical decompression of left temporal and frontal arachnoid cysts, with abnormal test values returning to those seen in a normal reference group after surgery.93 Hund-Georgiadis et al used functional MR imaging to study language dominance and regional temporal anatomy in five right-handed patients with large left-sided arachnoid cysts. Four were left-dominant for language, despite the large ipsilateral cysts, and the fifth showed mixed dominance.94 Positron emission tomography (PET) studies in a similar group of four right-handed patients with left-sided cysts showed no right-sided language activity. However, PET/CT studies in a 51-year-old with a very large right frontal arachnoid cyst demonstrated normal cortical metabolism adjacent to the cyst, but the right cortical motor pathways were reorganized.89 Single-photon emission computed tomography (SPECT) studies of three children with middle fossa arachnoid cysts demonstrated both local and contralateral perfusion defects that resolved after surgical intervention.95 SPECT and cognitive improvement after surgical cyst treatment has also been reported.96
Nonneurologic consequences of cysts have also been reported. Pressure from a cyst may thin the bone adjacent to it, rarely even to the point of incompetence, with CSF leaks occurring to the middle ear,97 scalp98 and nasal cavity.99–101
13.2.2 Incidence and Epidemiology
After a review of nearly 12,000 pediatric MR imaging studies at a single institution, Al-Holou et al reported the MR imaging incidence of arachnoid cysts in children to be 2.6%. The male incidence was 3.8%, versus 1.8% for the female incidence. This male predominance persisted throughout all pediatric age ranges.102 Males also had statistically larger cysts than females on average. There was no increase in the incidence throughout childhood—that is, new cases were not detected more commonly at older ages. Middle fossa cysts accounted for 45% of all cysts and were twice as common on the left side as on the right. About one-third of cysts were located in the posterior fossa, with the majority posterior to the cerebellum and 6% in the cerebellopontine angle (▶ Fig. 13.2).102 Contrasting with the findings in earlier, smaller studies, there was no gender difference between cyst sidedness.79,103 Earlier estimates in the literature, also from smaller samples, noted incidence rates from 0.3 to 1.7%, with the study patients mostly adults.104–106 Most incidence studies have similarly identified an increased incidence of cysts in males and a left-sided predominance, particularly for middle fossa arachnoid cysts. Using the same methodology as in the aforementioned pediatric study, Al-Holou et al reported a 1.4% adult incidence of arachnoid cysts drawn from a database of more than 48,000 MR imaging studies, with a male predominance (1.8% vs. 1.1%) There was no change in incidence by age. Middle fossa cysts (34%) and retrocerebellar cysts (17%) were seen most commonly.90
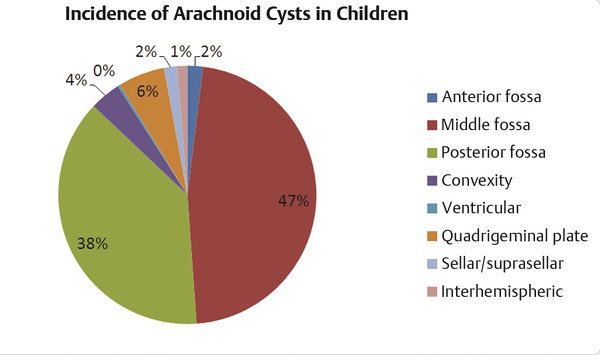
Fig. 13.2 The incidence of arachnoid cysts in 309 children drawn from a sample of 11,738 consecutive MR imaging studies.
(Modified from Al-Holou WN, Yew AY, Boomsaad ZE, Garton HJ, Muraszko KM, Maher CO. Prevalence and natural history of arachnoid cysts in children. J Neurosurg Pediatr 2010;5(6):578–585.)
Arachnoid cysts have also been reported in multiple other sites, including within the sella turcica,107,108 within the ventricular system108–110 and between the cerebral hemispheres.111 Suprasellar, quadrigeminal plate, ambient cistern, cerebellopontine, and retrocerebellar arachnoid cysts are also seen.
Middle fossa cysts have been classified by Galassi et al.112 Type 1 cysts are small and lens-shaped, are located at the proximal sylvian fissure, and do not produce any midline shift. Galassi et al reported that these cysts appear to fill with spinally injected contrast media. Type 2 cysts are more rectangular in shape and extend in the sylvian fissure to border the insula. Minimal midline shift may be present. Type 3 lesions involve the entire sylvian fissure, with thinning of the adjacent bone and more obvious midline shift, and they do not appear to communicate with the subarachnoid space on contrast injection. Of the middle fossa cysts in children reported by Al-Holou et al, 68% were type 1, 15% were type 2, and 17% were type 3.102 In most case series, type 1 lesions are less likely to be treated than type 2 or 3 lesions.
No clear ethnic or racial trends have been reported for arachnoid cysts. Familial occurrence has been reported.113 Arachnoid cysts have been associated with a myriad of conditions in case reports, but given their relatively common occurrence, these associations are more likely to be coincident than causal. However, two conditions are more firmly linked to arachnoid cysts. Autosomal-dominant polycystic kidney disease (ADPKD) is associated with an increased incidence of arachnoid cysts. Schievink et al reported an 8.1% incidence of arachnoid cysts in a group of 247 patients with ADPKD. There was also a higher incidence of cystic liver disease in the patients with ADPKD and arachnoid cysts than in those without. However, no patient in this series required treatment for an arachnoid cyst.114 Additionally, glutaric aciduria type 1 is associated with bilateral middle fossa arachnoid cysts. Patients with this disorder may be at increased risk for harm during surgical procedures.115–118
13.2.3 Natural History
Size
Most arachnoid cysts do not expand after initial radiographic diagnosis. In a study of 111 pediatric patients with a mean follow-up of 3.5 years and serial MR imaging, 87 cysts remained stable in size, 11 increased in size, and 13 decreased in size. Only 3 patients in this series became symptomatic. All patients whose cysts expanded were less than 4 years of age at initial diagnosis, and follow-up was similar for those younger and older than 4 years.102 Among adult patients studied by the same group, of 213 cysts followed over a mean of 3.3 years, 5 (2.3%) increased in size, while 2 (1%) decreased in size.90 A number of other studies have identified the connection between young age at diagnosis and the possibility of cyst expansion. Rao et al reported 2 children, 4 years of age or younger, who developed symptomatic expansion. In reviewing the pediatric literature to 2005, they identified 3 additional cases of radiographically documented symptomatic expansion in children who, they reported, were ages 8 months, 7 years, and 6 months.119,120 Radiographic resolution has also been documented on follow-up imaging.121–123
Intracystic Hemorrhage and Subdural Hygroma
Rarely, arachnoid cysts may rupture and present with a subdural CSF fluid collection. This is often associated with either modest or more dramatic subdural hemorrhage, presumably from the tearing of bridging veins associated with the cyst wall. Rupture has been reported to occur both after trauma and spontaneously.124–126 Radiographic resolution has been reported after rupture127 and has been seen in the author’s practice even without surgical intervention. Cress et al reported hemorrhage in 14 of 232 patients (6%) identified through an imaging database at a single institution. Using a case–control methodology, matching the patients for age, sex, side, and anatomical location with patients who had unruptured arachnoid cysts, the authors noted that children with rupture at presentation were more likely to have symptoms of intracranial hypertension and to have midline shift associated with their cysts. Cyst size larger than 5 cm in any dimension and prior head injury within 30 days were seen with statistically much greater frequency in the cohort with rupture, but altitude of residence was not different. Of the 14 patients with rupture, 10 were managed surgically. All patients recovered fully, but 14% required permanent CSF shunt placement.128 Other studies have reported a lower incidence of hemorrhage. Al-Holou et al reported 1 patient presenting with hemorrhage among 309 children (0.3%) with arachnoid cysts identified on MR imaging.102 Wester and Helland reported 11 of 246 adult and pediatric patients (4.6%) presenting with hemorrhage. All 11 patients underwent surgical treatment and recovered without postoperative morbidity. In this later study, cyst size was not noted to be a risk factor.129 Parsch et al, in a largely adult series, noted that 2 of 96 incidentally identified arachnoid cysts drawn from 11,487 imaged patients presented with hemorrhage.130
13.2.4 Clinical Presentation
Many arachnoid cysts are diagnosed incidentally when MR imaging is performed for an indication likely to be unrelated to their presence. In the University of Michigan pediatric series, only 6.8% of patients with radiographically identified arachnoid cysts were felt to be symptomatic.102 However, among the patients deemed symptomatic, the clinical manifestations related either to generalized increased ICP or to focal symptoms resulting from displacement or compression of specific structures by the arachnoid cyst. In symptomatic infants, there is often associated hydrocephalus, and the presentation of progressive macrocephaly, split sutures, and a Parinaud syndrome can occur. In older children, headaches, nausea and vomiting, lethargy, sixth nerve palsy, and papilledema can all occur. In the middle fossa, a cyst can cause focal prominence of the overlying bone and distortion of the adjacent orbit with proptosis. Suprasellar arachnoid cysts can produce endocrinopathy related to deformity of the pituitary axis and visual disturbance from displacement of the optic apparatus. Quadrigeminal plate arachnoid cysts most frequently produce hydrocephalus but may cause a Parinaud syndrome from local deformation of the tectum. Cerebellopontine angle cysts have been associated with nystagmus, facial weakness, hearing loss, and tinnitus.131 Given the above descriptions, the clinician is faced with the task of determining, for an individual patient, whether there is a causal relationship between reported complaints and the identified cyst. Information regarding the sensitivity and specificity of common features obtained from the history and physical examination for predicting the potential for future harm or a positive response to treatment is not widely available. This is especially true for common protean symptoms like headache.
Headaches
In most pediatric series of symptomatic patients, headaches are among the most common presenting symptoms.102,132–138 The headaches that are associated with arachnoid cysts in the absence of overt hydrocephalus have not been specifically characterized in the literature. Unlike in other neurosurgical conditions, such Chiari 1 malformation, in which there appears to be a correlation between the specifics of the headache and the likelihood of response to surgical therapy, no similar specificity has been demonstrated for arachnoid cysts. One can speculate that the pain arises from dural compression or displacement of the dura, given its rich sensory innervation, although focal episodic pain is more likely to be migrainous in an otherwise neurologically normal child. However, why some patients with cysts that cause mass effect on adjacent bone or midline shift have headaches and others do not is not known. One study has looked at the relationship between intracranial hypertension, cyst size, and clinical symptoms. Di Rocco et al reported on 11 children with middle fossa arachnoid cysts who underwent ICP monitoring. The ICP was observed to be normal (< 10 mm Hg, author’s definition) in 3 children with smaller, Galassi type 1 cysts, although 2 of these 3 had headaches. Of 7 patients who had larger, Galassi type 2 cysts, none had headaches. Half of these children had ICPs more consistently about 10 mm Hg, half had ICPs below. Two patients with larger, type 3 cysts had significant ICP elevations, one with headache.139 Migraine headaches in children are common, with a prevalence of 2.7 to 10.6%140 Given that arachnoid cysts are also relatively common, a significant number of children will present with both entities as a matter of chance.
Epilepsy
In surgical series, seizures occur in 20 to 34% of patients with arachnoid cysts.132,134,136 However, this is almost certainly a selection bias, because the incidence in nonsurgical series is considerably lower. In the previously noted MR imaging study of the incidence of arachnoid cysts, Al-Holou et al reported that there was a concern for seizures in 16% of children undergoing MR imaging studies in whom an arachnoid cyst was present.102 Presumably, the actual incidence of epilepsy was lower. The relationship between arachnoid cysts and seizures is not straightforward. Although one might postulate a global effect of an arachnoid cyst, increasing the propensity for epilepsy, it is more likely that the effect of the cyst would be felt most on the adjacent tissue. A majority of arachnoid cysts present in the temporal fossa. The temporal lobe is more prone to epilepsy than other regions of the brain. So it might be expected that complex partial epilepsy would be the most common type in patients with arachnoid cysts. Arroyo and Santamaria identified a 2% incidence of arachnoid cysts in a clinic for adults with refractory epilepsy. However, only 3 of 12 patients with middle fossa cysts had temporal lobe epilepsy. Interestingly, about 25% of the patients with arachnoid cysts in this series were found to have cortical dysplasias remote from the arachnoid cyst.141 Similarly, Yalçin reported seizure diagnoses in 21 patients with arachnoid cysts, mostly in the middle fossa, drawn from a clinical population of 612. Eleven patients had generalized seizures, and 5 had focal nontemporal epilepsy. Four patients had temporal complex partial events, but of these, only one had an ipsilateral cyst.142 Arai et al assessed with preoperative electroencephalography (EEG) 77 patients with middle fossa arachnoid cysts, who were subsequently treated with cyst–peritoneal shunt placement; 54% had EEG abnormalities and 34% had documented seizures, of whom 16 had generalized seizures, 8 had simple partial seizures, and 2 had complex partial seizures. After shunt treatment with a high rate of cyst decompression, 71% of the observed postoperative EEGs were either unchanged or worse. Of 26 patients, 21 required ongoing medical therapy for seizures, 1 had resolution of the epilepsy, and 4 required significant additional medications or alternative surgery or died as a result of their epilepsy.133 Conversely, multiple surgical series, reviewed below, do show considerable improvements in seizure outcomes, more strongly implying a causal connection.
Developmental Delay and Cognitive Deficits
Surgical series report cognitive disorders and developmental delay in up to a 25% of patients undergoing treatment.132 In some cases, these are believed to be due to the arachnoid cyst.143 As noted previously, pediatric patients with arachnoid cysts have been demonstrated to have abnormal SPECT studies95,144 and PET/CT studies in the brain adjacent to the arachnoid cyst.145 Neuropsychometric testing in 55 adults conducted pre- and postoperatively and compared with testing in a group of healthy volunteers demonstrated both significantly worse performance at baseline and postoperative improvement in measures of visual retention and of attention and interference, among other parameters tested.146 Similar improvements in verbal memory in adult patients after cyst decompression have been reported by the same group.147 However, pediatric case series consistently report little improvement in cognitive delay in patients undergoing cyst decompression, even when other symptoms, such as ICP-related headaches and progressive macrocephaly, are improved.133,134
Suprasellar Cysts and Neuroendocrinologic Disturbances
Suprasellar cysts and large middle fossa cysts that encroach on the hypothalamic–pituitary axis unsurprisingly can produce significant endocrinologic disturbances. Central precocious puberty, inadequate growth hormone production, hypothyroidism, amenorrhea, low plasma testosterone, diabetes insipidus, and the syndrome of inappropriate antidiuretic hormone secretion have all been reported.91,92,148,149 Endocrinopathy is frequent enough in patients with suprasellar cysts to recommend screening for all such patients.
The bobble-head doll syndrome, a movement disorder characterized by head bobbing in both the sagittal and coronal planes, usually at a frequency of 2 to 3 Hz, is associated with lesions of the hypothalamus and third ventricle. It has also been reported with suprasellar arachnoid cysts.150–152 In these case reports, cyst treatment is associated with resolution of the symptoms. El-Ghandour, reporting a case series of 25 children with suprasellar arachnoid cysts, noted that all had hydrocephalus, 5had developmental delay, 1 had visual impairment, and 1 had precocious puberty. Two patients had the aforementioned bobble-head doll syndrome.151
Intrasellar arachnoid cysts have also been reported but are rarely diagnosed in children.153
Cerebellopontine Angle Cysts
Cerebellopontine angle arachnoid cysts account for about 15% of all cysts identified in children.90,138 In adult patients, cerebellopontine angle cysts are more likely to be symptomatic than arachnoid cysts generally, but this is not clearly so in children.90,102 Symptoms reported in children include headaches, vomiting, facial weakness, hearing loss, tinnitus, and ataxia.131
13.2.5 Radiology
CT and particularly MR imaging are the principal radiographic tools for diagnosis. Although MR imaging is likely the more useful, CT studies can still play an important role in sorting through the differential diagnoses of arachnoid cysts in certain locations and can be an important tool in identifying the rare coincident intracystic or subdural hemorrhage. On CT, cysts without hemorrhage should be identical in density to CSF. MR imaging sequences also can help to demonstrate subdural hematoma in arachnoid cysts.154 A pitfall is that fat is similar enough in intensity to CSF on CT scans that certain lesions, such as cerebellopontine angle dermoid or epidermoid cysts, cannot be readily distinguished. For this latter diagnosis, even standard T1- and T2-weighted MR imaging may not be helpful. Diffusion-weighted images, however, readily differentiate the hyperintense dermoid cyst from the hypointense arachnoid cyst and should be part of the evaluation of intracranial cystic lesions.155 Conversely, CT can be very helpful in identifying calcium in suprasellar lesions, which is sometimes hard to discern on MR imaging sequences. Calcium in a cyst wall makes it much more likely for a lesion to be a craniopharyngioma than a suprasellar arachnoid cyst.
With the exception of cysts in the intraventricular location, MR imaging studies should demonstrate that the arachnoid cyst is extra-axial. Particularly, there should be a gray matter boundary to the cyst on the brain side. The author has cared for a child referred with the diagnosis of an expanding arachnoid cyst. On inspection, the lesion proved to have a white matter boundary and was instead an expanding tumor cyst. ▶ Table 13.1 lists entities in the radiographic differential diagnosis by anatomical location.
Suprasellar arachnoid cysts frequently produce hydrocephalus via obstruction at the level of the foramen of Monro. The third ventricle is compressed and displaced posteriorly. However, because the cyst itself is of CSF intensity, on axial imaging, particularly with CT, it can be mistaken for the third ventricle, leading to a mistaken impression of an aqueductal obstruction. In axial imaging, the foramen of Monro is typically bowed anterolaterally by the expanding extra-axial suprasellar arachnoid cyst. Sagittal plain imaging is usually diagnostic (▶ Fig. 13.3).
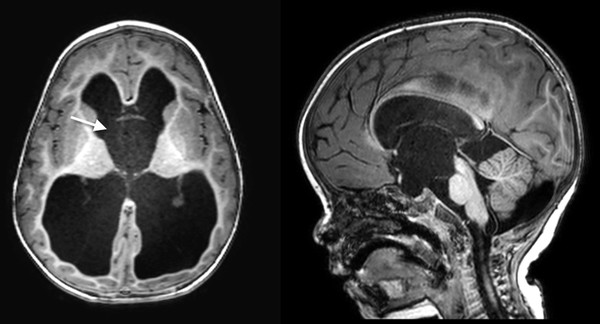
Fig. 13.3 Suprasellar arachnoid cyst. Note the convex appearance of the region of the foramen of Monro.









