Fig. 1.1
(a) Traditional concept that the infarct core is surrounded by an enveloping penumbra. (b) Alternative concept that regions corresponding to core and penumbra may be heterogeneous
Ischemic heterogeneity is also demonstrated by recent PET studies. Baron, Donnan, Heiss, and others have re-explored the penumbra concept using PET markers [25]. The benzodiazepine 11C-flumazenil binds to the intact GABA-A receptors of presumably intact neural tissue. In acute patients, PET documented areas of decreased 11C-flumazenil uptake went on to show infarctions, while areas of relative hypoperfusion exhibited a variable fate, even though 11C-flumazenil binding was within a normal range [26]. Another tracer, 18F-fluoromisonidazole, marks hypoxic regions and such images provide additional support for the heterogeneity of core and penumbral regions [27, 28]. Taken together, these experimental and clinical studies support a model of ischemia which includes heterogeneity of both core and penumbra. For example, as shown in Fig. 1.1b, the central core of critically low blood flow could be surrounded by patches of penumbral flow. Some of these penumbral patches, in turn, may also include zones of very low or core-type blood flow.
Incomplete Reperfusion
Despite the apparent restoration of blood flow through arteries supplying the penumbra, there may be protracted and heterogeneous penumbral cell injury, for unclear reasons. One possibility is that variable areas of brain receive penumbral levels of flow and the distribution of these areas in the brain is determined by variable degrees of collateralization from other blood vessels. Such collaterals generally connect in the pia between end-branches of the larger arteries or via the microvascular network. In contrast to the well-known sources of collateral flow among large arteries, an extensive network of capillary collateralization has been described in the mouse [29]. Whether such a rich source of microvascular collateral flow exists in humans remains to be shown.
Variability in blood-flow may influence the extent of vascular injury and inflammation during re-perfusion [30]. Throughout the time the microvessels suffer low flow, residual blood elements begin to coalesce (Fig. 1.2). Restoration of flow, if it comes too late, may exacerbate this process. Recent in vivo imaging studies of the cerebral microcirculation after transient MCAo demonstrate that re-perfusion is associated with accumulation of leukocytes along cerebral venule walls [31]. Such leukocyte accumulation can be blunted by hemodilution (albumin therapy) and with the improvement of venular re-perfusion, the final infarct volume is reduced [32]. Re-perfusion injury also worsens in a time-dependent manner to include platelet as well as leukocyte adhesion to vessel walls. This likely reflects a time-dependent increased expression of adhesive molecules which mediate platelet adhesion, such as P-selectin on cell surfaces of endothelial cells and platelets [33]. Other early markers of endothelial activation include E-selectin, ICAM-1, VCAM, and the urokinase plasminogen activator receptor (uPAR) [34, 35]. P-selectin in turn unleashes a cascade of inflammatory and thrombotic mechanisms [36]. Such data suggests vascular injury and subsequent microvascular occlusions may form a mechanism that underlies penumbral heterogeneity.
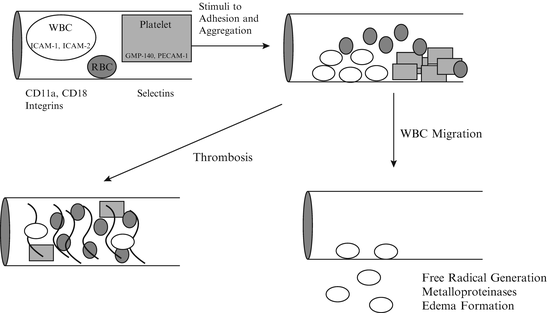
Fig. 1.2
Following occlusion, granulocytes leave the circulation to enter ischemic brain
Another reason for reperfusion failure may be the accumulation of thrombus in the distal microcirculation, proposed originally as the no-reflow phenomenon [37, 38]. Several groups have observed clot in the microcirculation distal to larger artery occlusion, although it can be challenging to eliminate the possibility of post-mortem clot formation. Elegant studies in the baboon striatum document a clear relationship between endothelial cell “activation,” by which the authors mean up-regulation of inflammatory markers that promote or signify thrombosis, and neuronal cell death [39]. Such data suggest that anti-thrombotic therapy should help stroke patients, but clinical trials of anti-thrombotic therapy, including abciximab, heparin, and heparinoid, all failed [40, 41]. It is not clear if these trials failed due to weak design (overly generous time window for enrollment, heterogeneous patient selection) or because distal microcirculatory thrombosis contributes relatively little in the patho-anatomic mechanism of penumbral flow failure. As a result of the clinical trial failures, alternative mechanisms must be considered.
Another reason for no-reflow into the penumbra after recanalization is downstream vasoconstriction. There are several mechanisms for downstream vasoconstriction. First, proximal large artery occlusion, such as tMCAo, releases a number of substances from the endothelium and from the in situ thrombus. Many of these substances act distally to cause smooth muscle cell contraction. It is not yet clear whether this phenomenon contributes meaningfully to outcome, but in vivo measurements do confirm distal changes in blood flow after arterial infusion of some of these substances [42, 43]. Second, it is now clear that astrocytes play a role in mediating regional rCBF, perhaps by directly stimulating arteriolar vasoconstriction and dilation [44, 45]. Data suggests that astrocytes communicate via gap junctions, and the calcium transients may mediate the propagation of signaling events downstream [46]. Third, direct muscular contraction could propagate down the arterial tree, from the site of proximal injury to the distal branch arterioles. While such propagating waves of vascular wall spasm have been documented in ex vivo preparations, a direct link to rCBF has not been proven.
A final and speculative reason for heterogeneous patterns of reflow is the variable distribution of effector pathways in the cortex and striatum. For example, there is a well-known pattern of selective vulnerability to global ischemia [47]. In areas of greater vulnerability, there is a much greater concentration of excitatory synapses compared to inhibitory synapses [48, 49]. Although largely unexplored, no doubt there are significant differences in the distribution of important effector systems such as arteriolar receptors for vasoconstrictive substances.
Regional Cerebral Blood Flow
The penumbra concept grew out of research directed at mapping the topography of cerebral blood flow. Traditional approaches to mapping rCBF (regional cerebral blood flow) include tracer studies (usually 14C-antipyrine), or hydrogen clearance, both of which are technically demanding. In fact, if one examines the data from the original studies that motivated the penumbra model, there are data points that fall outside the blood flow boundaries [1, 2]—it is possible that some electrodes were recording from islands of lower blood flow, while other electrodes were recording normal flow in the presumed penumbral shell. There is no way now to confirm this novel interpretation of that data, but studies will be needed to determine whether rCBF reduction is heterogeneous after MCAo.
Recent application of optical methods to the problem of in vivo blood flow has provided new insights into the effects of ischemia on microcirculation [50–53]. Via a cranial window, confocal laser microscopy has been used to visualize blood flow in individual cerebral vessels after infusion of fluorescently tagged blood cells in vessels up to 200 μm deep to the cortical surface. Moving erythrocytes are mapped, and from the time and distance moved, actual blood velocity is calculated directly. After 4-vessel occlusion, capillary flow was shown to be zero, which correlated with EEG flattening; flow returned after clamp release [54]. The application of a two-photon laser scanning microscopy (TPLSM) method by Kleinfeld and colleagues now allows visualization of flow in vessels up to 400 μm deep to the cortical surface and quantitation of flow in arterioles and veins, as well as capillaries [50, 51]. This methodology is compatible with experimental ischemia and has provided new insights into flow rearrangement following cortical microthrombosis [29]. The combination of classical immunohistochemical methods with in vivo imaging afford a unique opportunity to explore the mechanisms underlying penumbral injury. Both techniques will provide complementary evidence of blood flow changes and cellular injury in ischemic zones.
Until very recently, our concept of the penumbra simply included 2 zones, one surrounding the other, as shown in Fig. 1.1a. There are a number of problems with the classical penumbra concept. Despite the interesting data obtained by some groups, the vast majority of patients with focal ischemia do not exhibit such consistent patterns. Many more patients who respond to thrombolysis with a gratifying recovery do NOT show a pattern on PET or MRI consistent with a presumed core and a larger penumbra. It is increasingly clear that the penumbra concept should be expanded to include more complicated flow patterns [55]. For example, in Fig. 1.1b we illustrate a possible scenario in which a central core is surrounded by patches of penumbral flow, some of which include additional zones of very low, or core, flow. It is quite likely that variable areas of brain receive penumbral levels of flow and that the distribution of these areas in the brain is determined by variable degrees of collateralization from other blood vessels. Such collaterals generally connect in the pia between end-branches of the larger arteries. There is some limited pathological data to support this concept of “islands” of penumbral flow—the heterogeneous penumbra—near areas of complete infarction [55–57].
Support for the heterogeneous penumbra comes from a variety of sources. In reviewing some seminal papers, there is obvious heterogeneity in the published diagrams. For example, a diffusion MRI study from Stanford included a photograph of diffusion weighted images taken serially from one subject [16]. In Fig. 7 from that study, the image taken 1.5 h ischemia onset shows clear islands of abnormal signal in the striatum and overlying cortex. The islands begin to coalesce in the 2.5 h image, and are nearly confluent by 4 h. Similarly, PET images usually show a heterogeneous pattern of hypometabolism in the first hour or two following ischemia onset (see Fig. 1.3) [58].
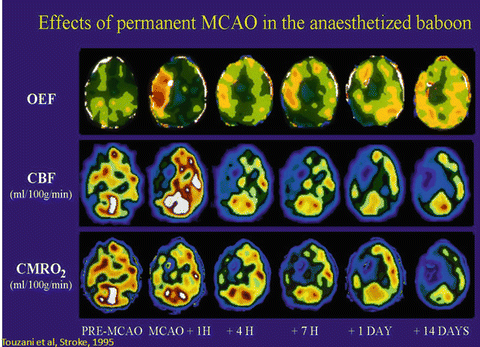
Fig 1.3
Effects of permanent MCAO in the anaesthetized baboon. (Reprinted with permission from Touzani et al. [58])
No matter what the spatial pattern of blood flow changes, it is clear that cells in penumbral regions do not survive indefinitely. The exact duration of survival is unknown, and likely it is different under differing circumstances. Cells in zones receiving 10–20 cc/100 g/min likely survive for hours, but the number of hours is not known. Many factors may reduce the time such marginally perfused cells might survive. For example, it is known that hyperthermia of 1 or 2 °C will accelerate cell death [59]. Elevations of serum glucose also accelerate cell death. Such marginal levels of blood flow do not support neuronal survival indefinitely, but could be sufficient to deliver neuroprotectants into the ischemic zone. Therefore, pending recanalization and restoration of blood flow, a number of therapies could be directed at idling neurons in the penumbra. Such neuroprotectants might salvage brain by preserving neurons until blood flow is restored. The possible mechanisms of neuroprotection include interrupting ischemic excitotoxicity, blocking apoptosis, and blocking the inflammatory response that follows ischemia.
Therapeutic Targets in the Penumbra
The elucidation of the excitotoxic cell death mechanism advanced stroke neurology considerably. An understanding of the biochemical events outlined below spawned a plethora of therapeutic trials. As of this writing (2013) no agent has yet proven successful in humans, despite considerable excitement from laboratory studies. Future successful neuroprotection depends upon further delineation of the steps in the ischemic cascade, and identifying candidate points for intervention.
The Ischemic Cascade and the Role of Glutamate
The sudden deprivation of oxygen and glucose sets into motion a set of events called the ischemic cascade. The use of the term cascade implies that ischemia proceeds in an orderly manner from the beginning of the cascade to the end. Alternatively, it is becoming clear that the several steps in the cascade occur simultaneously, rather than sequentially and in addition there appear to be multiple feed-forward, feed-back, and amplification steps (e.g., [60, 61]). A simple sketch of some of these phenomena is presented as Fig. 1.4.
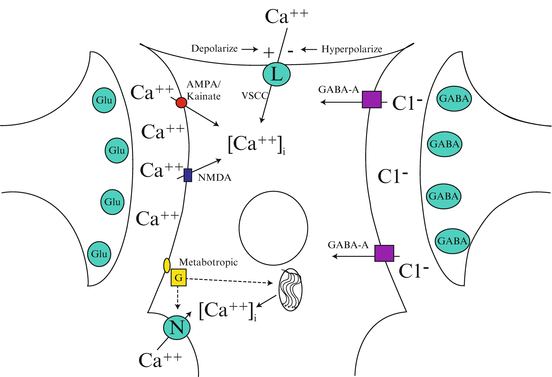
Fig. 1.4
Glutamate activation of any one of 3 channels results in depolarization and increased cytoplasmic calcium entry via ligand gated channels, voltage gated channels, and intra-cytoplasmic storage. γ-Aminobutyric acid-receptor activation leads to a hyperpolarizing influx of chloride that blocks voltage-gated calcium influx via the L-type voltage-sensitive calcium channel (VSCC). There may also be inhibition of the N-type calcium channel
Glutamate exposure is associated with cell death and degeneration in cell culture [62–65] and glutamate receptor antagonists protect cultured cells during exposure to glutamate, hypoglycemia and hypoxia [66]. At least 3 glutamate receptor sub-types are identified based on ligand binding studies: N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazoleproionate (AMPA)-kainate, and metabotropic [67, 68]. Of these subtypes, the NMDA receptor appears to be critical in mediating the effects of ischemia, although interest in AMPA/kainate receptors continues, especially in studies of global ischemia [69, 70] and the role of the metabotropic receptor sub-type remains unclear [71, 72].
Depolarization of the post-synaptic cell occurs in response to application of glutamate, and appears to be a necessary step in the sequence of events leading to cell death [63, 64]. During excitation of the post-synaptic membrane, there is an influx of sodium, chloride, calcium and water into the cell [66, 73]. Inflow of ions and water leads to edema, and if severe or prolonged, such edema may lead to cell lysis and death. Glutamate does not cause edema or lysis of mature neurons in culture unless sodium and/or chloride are present in the culture medium [73]. Inflow of calcium may lead to delayed neuronal death through unknown mechanisms after comparatively brief exposure to ischemia or excitotoxins [74]. There is some evidence that this delayed cell death may be mediated through apoptotic mechanisms [75–78]. This effect persists in culture if sodium is not present in the medium but is blocked by MG++ or by removing calcium from the medium [64].
During ischemia, intracellular calcium concentrations increase through mechanisms other than the ligand-gated channels described above. Some calcium channels are voltage gated, and depolarization to a membrane voltage that opens some of these channels may be a critical determinant of calcium influx [64, 79]. However, since calcium appears to enter the cell through the NMDA receptor itself, ligand-gated influx appears to continue even in voltage clamped cells [80]. Nevertheless, it seems reasonable to suspect that prevention of glutamate-stimulated depolarization ought to prevent some of the early cellular edema due to sodium, chloride and water movement, and some of the calcium flow that leads to delayed toxicity. In support of this expectation, it was observed that hyperpolarization reduced or blocked calcium inflow into neurons [81] and reduced the probability of discharge [82]. Other routes of influx include the release of calcium from intracytoplasmic stores (mediated in part via metabotropic receptors linked to protein kinase C) and loss of ATP dependent calcium extrusion mechanisms [83]. Also, with membrane damage there is influx of calcium down an electrochemical gradient [84].
There is now a growing consensus that the increase in intracellular calcium sets into motion a variety of events that lead to cell death, most especially the activation of cytosolic phospolipases, and proteases [85–87]. Proteases may play a central role in activating programmed cell death; phospholipases may be critically involved in further excitotoxin release, and the generation of reactive oxygen species; as a direct result of early glutamate mediated increases in intracellular calcium a vicious cycle is created that causes spread of the core zone of infarction.
Several direct glutamate antagonists (MK-801, CGS-19755, and dextrophan) and indirect agents that bind at the NMDA glycine site (ACEA1021, felbamate, GV150526a) appear to protect brain during focal ischemia [68, 88–92]. The direct agents resemble dissociative anesthetics such as ketamine and phencyclidine and humans experience significant side effects during treatment with NMDA antagonists that ended development efforts for some agents. An alternative strategy for brain protection during cerebral ischemia is to use agents that block the effect of excitotoxins without causing such severe side effects. Agonists of the GABA receptor have been studied for the same purpose, but as with direct glutamate antagonists, no benefit has yet been shown in clinical trial [93–95]. Magnesium is a potent inhibitor of the NMDA receptor that has shown benefit in experimental models; human trials continue [96–101].
The Role of Apoptosis and Necrosis
Despite the obvious difference in blood flow between the core and the penumbra, the mechanisms of cell death in the 2 regions are not fully known. Necrosis is obviously one mechanism for cell death in both the penumbra and the core. During necrosis the cell initially swells, then shrinks and can be observed as a small, pyknotic form on sections [102]. Finally, microglia and macrophages remove the debris of the dead cell. If necrosis includes the adjacent glia and structural matrix, a cyst is formed and the process is termed pannecrosis. In addition, apoptosis, or programmed cell death, has been observed in ischemic brain [78]. In this type of cell death, ischemia is thought to activate “suicide” proteins that are latent in all cells. These proteins are normally expressed during embryogenesis and enable the organism to remove cells that will not be needed during further development. The morphometry of apoptosis is quite different from necrosis, and special techniques are available to study the 2 forms of cell death [103]. The role of apoptotic cell death in the core and in the penumbra is not known.
Injuries such as hypoglycemia, anoxia and excitotoxin exposure kill cells via necrosis. Necrotic cell death is clearly associated with elevations of intracellular calcium, which has been demonstrated in culture, brain slices and intact brain. The histopathologic sequence of cellular changes leading to necrosis has been documented [102, 104]. After focal or global ischemia swollen, eosinophilic neurons appear within 2–8 h. Pyknotic, shrunken neurons appear within 24–36 h. Phagocytosis of neurons, astrocytes and surrounding matrix occurs over days. Once the necrotic cell death pathway begins, it probably cannot be interrupted easily. In areas of severe ischemia (core) a cyst is formed after phagocytosis and removal of all brain elements. In surrounding brain (penumbra) some cells are removed and astrocytes may proliferate, leaving a zone of “incomplete” infarction that is depleted of neurons but not cystic. The neurological consequences of this incomplete infarction are unknown.
Injuries such as free radical excess or mild ischemia in cell culture cause programmed cell death via a series of events that may represent apoptosis. The initial events are shrinkage of the nucleus and cytoplasm, chromatin condensation followed by nuclear fragmentation, and the separation of cell membrane protuberances (blebs). A hallmark is the cleavage of DNA by endonucleases into segments of non-random length, which results in “laddering” on DNA isolation gels. Ultimately the cell separates into small, pyknotic bodies that are phagoctytosed by cells resident in the tissue. There is little or no inflammatory infiltrate associated with apoptosis. During some phases, it is difficult to distinguish apoptotic from necrotic cells using morphologic criteria [3]. Following focal cerebral injury, including middle cerebral artery occlusion, apoptotic-like changes can be documented in brain [78, 105–107]. Specifically, focal ischemia is associated with DNA nicking as documented with labels specific for free DNA strands, non-random DNA fragmentation as documented by the “laddering” phenomenon on DNA extraction gels, and ultrastructural findings consistent with apoptosis. It is not clear if apoptotic-like cell death occurs in the core, the penumbra or both. It is also not clear whether this pathway can be interrupted. Of most concern, it is not clear whether apoptotic-like cell death occurs separately from necrotic death, or whether the two phenomena represent different manifestations of one underlying cell death process. It is very clear that necrotic and apoptotic appearing cells can be found in the same regions of brain at the same time [108].
The specific pathways involved in apoptotic CNS cell death remain to be elucidated. In some circumstances, the cell death mediator caspase-3 appears to be involved, and preliminary studies with caspase inhibitors show neuroprotection [109]. There are caspase-independent pathways as well that seem to involve an apoptotic-like mechanism [110, 111]. Other events, including caspase-9 activation and Cytochrome-c release play a role in mediating ischemic cell death [110, 112–114].
The Role of Granuolcytes
Restoration of arterial blood flow after several hours of occlusion may not result in complete tissue reperfusion, the so-called no-reflow phenomenon described above [37, 38, 115]. The no-reflow effect does not occur if white blood cells are removed from the circulation [115]. Granulocytes may adhere to ischemic endothelium, blocking capillaries and stimulating platelet aggregation and formation of micro-thrombi, as illustrated in Fig. 1.2. The receptor complex on the granulocyte that mediates adherence is composed of the integrins CD18/CD11 (for review see [116, 117]). This complex binds to the intercellular adhesion molecule (ICAM) on the endothelial cell.
In addition to mediating the no-reflow phenomenon, granulocytes may have direct toxic effects in the brain. After adhesion, granulocytes migrate into the brain (diapedesis), and can be observed in the peri-ischemic zone within hours of permanent or transient arterial occlusion [118, 119]. Once in the brain granulocytes release phagocytic chemotactic factors as well as cytokines that may promote cellular destruction. Granulocytes also release enzymes that lead to the formation of free radicals, which leads to an increase in hypochlorous acid and chloramines [120]. These compounds then activate granulocytic serine proteases and metalloproteinases which together begin to destroy surrounding tissue. These events proceed independently of the energy status of the brain cells and may be augmented by early reperfusion, the reperfusion injury syndrome. Granulocytes have been detected in the penumbra in multiple animal models [121, 122]. There is very little evidence of reperfusion injury in humans after cerebral thrombolysis, however.
Therapies targeted at granulocytes appeared promising in animal models [123]. Unfortunately, a human trial was negative, and in fact patients treated may have suffered even worse outcomes [124]. There may be many reasons for this trial failure, but importantly, the antibody itself may have induced a host immune response that worsened stroke severity [125]. This treatment failure emphasizes the need for careful translation of animal results to human clinical trials as there may be unforeseen or unintended consequences from the putative neuroprotectant.
The Role of Macrophages
The involvement of monocytes/macrophages in ischemic brain following stroke has been well-characterized [126]. These inflammatory cells contribute primarily to phagocyte-mediated tissue debridement and scar formation days to weeks after stroke and are involved with post-ischemic hypoperfusion, neuronal dysfunction and blood-brain barrier permeability [127–129]. Maximal numbers of inflammatory cells accumulate in the ischemic area at different times. Accumulation of polymorphonuclear leukocytes is maximal at 24–72 h followed by monocytes and macrophages at about 7–16 days. After ischemia, macrophages arise from 2 sources: parenchymal microglial cells and the blood-borne monocytes [130, 131]. Microglial cells may develop into amoeboid microglia (brain macrophages) when neurons are lethally injured [132]. Both the activated microglial cells and the monocyte-derived macrophages are capable of inducing neuronal cell death and neurological dysfunction through secretion of cytotoxic factors [133]. To date, treatment strategies targeting macrophages have not succeeded [134, 135].
The Role of Microvessels
The extent of blood flow in the penumbra correlates strongly with the number and volume fraction of microvessels [136–138]. Volume density relates to the maximum volume of blood that could be flowing within the microvascular system. Changes in volume density and diameter are important descriptors of microvessel hypertrophy or fusion. The resistance to blood flow through microvessel networks is a function of microvessel diameter as well as microvessel length, density, and branching. The rate of blood-brain exchange is proportional to microvessel surface area and length density [129, 139, 140]. Beginning hours after stroke in rodents, genes related to angiogenesis up-regulate, producing both message and translated proteins [141]. Vascular endothelial growth factor (VEGF) is expressed in neurons for days, and for up to a week in pial cells. VEGF expression is apparent in astrocytes for up to 2 weeks after stroke [142–144]. In humans, the angiogenic platelet-derived growth factor (PDGF) message can be detected around cystic infarction for weeks after stroke [145]. These angiogenic signals may represent an attempt at neuroprotection: another angiogenic peptide, fibroblast growth factor (FGF), is neuroprotective in culture models of excitotoxic cell death, and VEGF could possess similar properties [146]. Alternatively, perhaps the brain utilizes VEGF and other angiogenic factors after stroke as part of a repair process. If there were brain regeneration after injury, new tissue would obviously require new vasculature. It is intriguing to note that VEGF, in addition to mediating vasculogenesis during development, may play a role in neuronal cell migration during embryogenesis [147].
As a third alternative, angiogenic factors have other properties, such as opening the blood brain barrier (BBB), which might be important in the removal of necrotic tissue; in this case, VEGF might have nothing to do with protection or recovery [148, 149]. In fact, this “clean-up” hypothesis is strongly suggested by recent data [129, 150]. It appears the brain utilizes the angiogenic-signaling cascade to open capillaries and synthesize microvessels needed for macrophage infiltration for the destruction/removal of necrotic brain. After formation of the cystic cavity, i.e., pannecrosis, the microvessels are no longer needed and are therefore degraded. In support of this, the majority of VEGF up-regulation was localized to microglia and macrophages [144]. Based on considerations of the ‘clean-up’ hypothesis, treatment strategies targeting angiogenesis have not proceeded [151].
Combinatorial Neuroprotection
As outlined above, there are a variety of interacting events proceeding simultaneously during ischemia and it now seems unlikely that a single agent will prove insufficient to salvage most of the ischemic brain in most patients [152]. On the other hand, multiple agents targeted at different receptors or events in the ischemic cascade may cooperatively improve outcome. Attempts have been made to design a combination of neuroprotective agents, with some success, but it is difficult to predict the doses of the two agents to use. Furthermore, the combination may manifest benefit as increased potency compared to the single agents or it may lengthen the treatment delay time window, or both (see Chap. 3). For example, when both rt-PA and a granulocyte inhibitor were given 30 min after ischemia onset, each was effective, as was the combination [123, 153]. After a treatment delay of 90 min t-PA and the combination were effective, but there was no synergistic benefit. After a delay of 180 min, t-PA was not effective when used alone, nor when combined with anti-ICAM given 5 or 175 min after ischemia. In a follow-up study, the same group studied longer time intervals [153]. In this study, t-PA given 2 h after embolization was not effective, but the combination of anti-ICAM 15 min after and t-PA 2 h after embolization was quite effective. Again, the dose of t-PA alone was not increased to see whether a maximal dose of the single agent was equipotent to the combination. However, this is highly unlikely because the dose of t-PA used, 3 mg/kg, is known to thrombolyse the majority of the injected emboli. Therefore, this study suggests that the use of the two agents conferred a benefit that could not be obtained from higher doses of either agent alone, i.e., synergism.
Hypothermia is the most potent neuroprotectant every studied (see Chap. 3). Mild hypothermia shows significant benefit after focal or global cerebral ischemia in animals and humans by reducing metabolic demand, limiting the inflammatory response to ischemia, reducing excitotoxin release and free radical generation, and limiting blood-brain barrier breakdown [154, 155]. Hypothermia treatment in ischemic rat animal models has been reviewed extensively in the literature [156] and improved neurologic outcomes have been demonstrated [157]. In models of global ischemia, the effect of hypothermia vastly depends on ischemia duration and time between ischemia and onset of hypothermia.
The optimal duration and depth of hypothermia remain controversial [158]. Data are accumulating to support the beneficial effect and safety of mild (34–35 °C) hypothermia in comparison to moderate/severe hypothermia that is often accompanied by increased systemic adverse effects [159–161]. The duration of hypothermia has been studied over a variety of maintenance periods (1–72 h), and substantial protection has been shown even at the shortest duration of 1 h [162]. The benefit of hypothermia in reducing infarct size and neuronal death (both cortical and striatal) has been shown to persist up to 6 months post-stroke [163]. This sustained benefit establishes that hypothermia may actually stop the ischemic cascade rather than simply delay inevitable damage.
In the treatment of stroke with hypothermia, rapid induction, precise temperature control, and ease of administration are critical requirements for the therapy. Cooling can be initiated through surface (skin) cooling or endovascular methods, such as catheter-based systems or intravenous infusions of chilled fluids [164]. Surface cooling using previous technologies may induce hypothermia slowly, however the latest generation of surface cooling devices has not been tried in awake stroke patients [165]. The average rate of cooling with surface methods ranged from 0.3 °C to 1.7 °C/h and can be significantly slower in patients with a high body mass index [166, 167]. Control around the target temperature was previously difficult with surface cooling. In the COOL AID study, target temperature overshoot was observed in all patients by as much as 5 °C [168]. More recent versions of surface cooling units may approach the cooling power of endovascular methods with greater control.
Other strategies for hypothermia induction include infusion of cold intravenous solutions, body cavity cooling with cold lavage, and extracorporeal cooling devices. These devices/methods have limitations. Body cavity cooling is either minimally effective or results in patient discomfort and complications. Gastric lavage with cold fluids can cause cramping and diarrhea. Bladder lavage is minimally effective. Use of cold intravenous solutions, combined with cooling water and air convection blankets, provides sufficient levels of hypothermia induction, and recent data suggest this approach can serve as a prelude to more definitive forms of cooling [169, 170]. Therefore, current protocols allow for hypothermia induction with 4 °C saline, followed by placement of an endovascular cooling catheter. Hypothermia with fluid loading alone is difficult to maintain for long periods of time as the patient is at risk for volume overload.
Endovascular cooling devices have been shown to reliably induce hypothermia, maintain target temperature, and provide stable re-warming without rebound temperature elevation [158, 171]. The ICTuS-L trial confirmed that hypothermia could be induced safely in patients receiving thrombolytic therapy with rt-PA [172]. The ICTuS-L trial also confirmed that very rapid and precisely controlled hypothermia can be induced in awake stroke patients with endovascular devices [171, 172]. The induction of hypothermia in stroke patients depends on the power of the cooling device, but the patient’s body mass index and age critically determine the speed of cooling [173]. While hypothermia may pose added risk, for example an increased risk of pneumonia, but further studies are need to confirm this [174].
References
1.
Astrup J, Symon L, Branston NM, Lassen NA. Cortical evoked potential and extracellular K + and H + at critical levels of brain ischemia. Stroke. 1977;8:51. PubMed PMID: 317.PubMed
2.
Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia: the ischemic penumbra. Stroke. 1981;12:723. PubMed PMID: 1323.PubMed
3.
Garcia JH, Liu KF, Ye ZR, Gutierrez JA. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke. 1997;28:2303. PubMed PMID: 3069.PubMed
4.
Heiss WD. Progress in cerebrovascular disease: flow thresholds of functional and morphological damage of brain tissue. Stroke. 1983;14:329. PubMed PMID: 314.PubMed
Related posts:
 Pilot and Preliminary Studies of Thrombolytic Therapy for Stroke
Combination of Intravenous t-PA and Endovascular Therapy
Thrombolytic Therapy for Acute Stroke and the Test of Time
Mechanical Revascularization with Embolectomy Devices
The Impact of Neuroimaging on Acute Stroke Treatment: Role of Computed Tomography
The Impact of Neuroimaging on Acute Stroke Treatment: Role of Magnetic Resonance Imaging
Pilot and Preliminary Studies of Thrombolytic Therapy for Stroke
Combination of Intravenous t-PA and Endovascular Therapy
Thrombolytic Therapy for Acute Stroke and the Test of Time
Mechanical Revascularization with Embolectomy Devices
The Impact of Neuroimaging on Acute Stroke Treatment: Role of Computed Tomography
The Impact of Neuroimaging on Acute Stroke Treatment: Role of Magnetic Resonance Imaging
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


