INTRODUCTION
It is yet to be discovered what induces the high vulnerability exhibited by specific groups of neurons in Alzheimer’s disease (AD), leading to their progressive degeneration. Vulnerable neurons are located in brain areas that are functionally linked to various forms and aspects of learning and memory, including the hippocampal formation, olfactory system and neocortex (for review see1). The parahippocampal regions are the earliest to be affected (Braak stages I and II). In particular, the entorhino-hippocampal circuit exhibits an early and significant neuropathology. Neuropathology in this region correlates significantly with degree of dementia. The hallmark lesions of the disease, most pronounced in the affected brain areas, are neurofibrillary tangles and amyloid deposits. Neurofibrillary tangles are intracellular aggregates of bundled paired helical filaments. The principal component of neurofibrillary tangles is hyperphosphorylated tau. The microtubule-associated group of tau proteins consists of alternatively spliced cytoplasmic proteins that bear either three or four microtubule-binding domains and co-assemble with tubulin onto microtubules, where they stabilize these organelles. As such, tau is implicated in numerous critical cellular processes, such as cell proliferation, neuronal maturation, process elongation and axonal transport. Thus, preceding neurofibrillary tangle formation, hyperphosphorylation of tau may interfere with crucial cellular and neuronal functions. The mechanism underlying hyperphosphorylation of tau in AD is not fully understood, as discussed below. Amyloid deposits are aggregates of β-amyloid (Aβ) peptides released from a large precursor termed amyloid precursor protein (APP) upon enzymatic cleavage. Upon increase production or fibrillogenic properties, Aβ undergoes conformational changes to form β-sheet structures that tend to form oligomers and subsequently insoluble aggregates. One form of amyloid aggregates is neuritic plaque, an extracellular core of amyloid fibrils surrounded by dystrophic dendrites, degenerating axons and reactive gliosis. Neuritic plaques are abundant in the molecular layer of the dentate gyrus of the hippocampus, the amygdala, the association cortices of the frontal, temporal and parietal lobes, and within certain deep brain nuclei that project to these regions. In addition to neuritic plaques, several other forms of Aβ aggregates exist, including abundant ‘diffuse plaques’ in the limbic and association cortices and even in non-conventionally affected regions, such as cerebellum. Increased fibrillogenesis and/or levels of Aβ is caused by either mutagenesis or ageing-linked unknown pathogenesis. In rare, familial, early onset form of AD (FAD), increased fibrillogenesis and/or levels of Aβ are caused by mutations in APP, presenilin-1 (PS1) and presenilin-2 (PS2). These mutations cause misprocessing and trafficking of APP by β-secretase or γ-secretase, the aspartyl protease of which PS is the catalytic core, yielding an increased amount and/or ratio of the fibrillogenic forms of Aβ (e.g. Aβ42), that consequently aggregate in the form of amyloid deposits. Both hallmark lesions are the result of misfolding and polymerization of otherwise soluble proteins. The progressive emergence of altered conformations of originally soluble proteins and subsequently insoluble aggregates, intra-and extracellularly, is associated with profound neuronal dysfunction and death.
FAMILIAL ALZHEIMER’S DISEASE-LINKED CENTRAL PLAYERS
Amyloid Precursor Proteins
Amyloid precursor proteins encompass a family of membraneanchored type I glycoproteins that are ubiquitously expressed and particularly abundant in the brain (for review see2). APP full length are 110-130kDa proteins that are synthesized in the endoplasmic reticulum. APP maps to chromosome 21 in humans and is transcribed to three isoforms, APP695, APP750 and APP77i with the former being predominantly expressed in neurons and the latter two in glia. APP770 is encoded by 18 exons, where exon 17 resembles the membrane-spanning domain. APP695 is lacking both exon 7 and exon 8, and is the most abundant APP transcript in the brain. APP75i (lacking exon 8), APP7i4 (lacking exon 7) and APP770 are expressed in the peripheral and central nervous system. Exon 7 encodes an amino acid fragment that exhibits substantial homology to the Kunitz-type serine protease inhibitor (KPI) while exon 8 shares some homology with the MRC OX-2 antigen. Two additional members of the APP family are amyloid precursor-like protein 1 and 2 (APLP1 and APLP2). They are truncated at the carboxyl terminus and lack the Aβ region. The APLPs map to chromosome 19 in humans. The APLP2 but not APLP1 contains a KPI domain. Nevertheless, neither mutations in APLP1 nor in APLP2 are causative of FAD, supporting the notion that Aβ plays a major role in the disease. Indeed, FAD-linked mutations introduce amino acid substitutions within or flanking the Aβ region of APP, around the cleavage sites of a-, β-and γ-secretase. APP undergoes a complex set of tightly linked trafficking and processing. From the endoplasmic reticulum, APP is transported to the plasma membrane through Golgi apparatus. However, only a small fraction of APP molecules reaches the plasma membrane. During this journey APP undergoes multi post-translation modifications of N – and O-glycosylation, ectodomain and cytoplasmic phosphorylation, and tyrosine sulphation. The presence of the YENPTY internalization motif near the C terminus of APP enables APP endocytosis shortly after it reaches the plasma membrane. Then APP is delivered to endosomes, and a fraction of endocytosed molecules is recycled to the cell surface. Some of internalized APP undergoes lysosomal degradation. During its journey, APP is subject to a complex processing by multiple enzymatic activities (Figure 49.1), most of which are around the 4 kDa region of the Aβ peptide (for review see1). In the nonamyloidogenic pathway, APP is cleaved by α-secretase at amino acid 16-17 of the Aβ region, preventing the formation of Aβ. In the amyloidogenic pathway, APP is cleaved by the β-secretase enzymatic activity at the carboxyl terminus of the Aβ region, followed by a cleavage by γ-secretase enzymatic complex at the amino terminus of the Aβ region, leading to the release of Aβ. Following shedding of APP by a- and β-secretase, the soluble product (sAPPa and sAPPβ) is released to the extracellular space, while a membrane-anchored fragment is subject to an intramembranous cleavage by γ-secretase, releasing APP carboxyl terminus fragments (AICD) into the cytoplasm. AICD can form a transcriptionally active complex with Fe65 and Tip60. While the role of AICD in regulation of gene transcription is yet to be confirmed, several genes are thought to be regulated by AICD: KAI1, the Aβ-degrading enzyme neprilysin, LRP1, and epidermal growth factor (EGF) receptor. Many adaptor proteins with phosphotyrosine-binding domains, including Fe65, Fe65L1, Fe65L2, Mint1 (i.e. X11), Mint2, Mint3, Dab1, and c-Jun N-terminal kinase (JNK)-interacting protein family members bind this motif and the flanking region. Overexpression of these adaptors reduces Aβ production and deposition in the brains of transgenic mice, suggesting that these adaptors play a role in regulation of APP processing (for review see3). Interestingly, Fe65 serves as a functional linker between APP and LRP in modulating endocytic APP trafficking and Aβ production. Mutations within the YENPTY endocytosis motif inhibit APP internalization and decrease Aβ production, suggesting that APP is processed to yield Aβ following internalization.
The phenotype of mice with homozygous ablation of APP appeared rather mild compared to presenilin-1 knockout phenotype. Some
Figure 49.1 Schematic presentation of APP processing in its amyloidogenic and nonamyloidogenic pathways. AICD = APP carboxyl terminus fragments Image generation by courtesy of Michael Demars
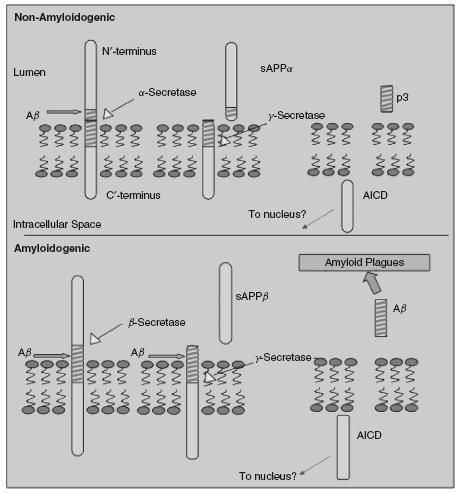
synaptic markers are thought to be reduced in APP-deficient mice, and this reduction correlates with deficits in learning and memory and in synaptic plasticity. APLP2 knockout mice appear normal, while APLP1 knockout exhibit some growth impairments. This mild phenotype might be due to partial functional redundancy among the family members. In support of this notion, APLP2 /APLP1 and APP/APLP2 double knockouts die right after birth, suggesting that APLP2 functionally overlaps with APP and APLP1. Examination of the central and peripheral nervous system of the double knockout mice confirmed significant deficits in synapse formation, deformed pre-and post-synaptic element distribution, reduced number of synaptic vesicles and aberrant localization of the choline transporter at neuromuscular junctions. Deficiency of all three App genes led to death shortly after birth, exhibiting cortical dysplasia suggestive of migration abnormalities of the neuroblasts and partial loss of cortical Cajal-Retzius cells. Taken together, these findings suggest that the APP gene family plays important roles in the development of the nervous system, such as synapse formation and regulation of neural progenitor cells.
Presenilins and y-Secretase
Presenilins (PS1 and PS2) are multipass membrane proteins that are predominantly localized to the endoplasmic reticulum. PS undergo endoproteolysis by an unknown enzymatic activity termed Presenilase, and the resulting amino-terminal and carboxy-terminal fragments that remain closely associated, are thought to be the active form of the protein. PS are thought to play numerous roles in the cell, but are best known for their central role as the catalytic core of the intramembranous, γ-secretase aspartyl protease that cleaves numerous membrane proteins, including APP. γ-secretase is a unique membrane protease complex. Presenilin 1,2 are thought to be the catalytic subunit of γ-secretase. PS contain two essential aspartate residues in putatively adjacent transmembrane domains (for review see4). Substitution of either of the two aspartate residues buried within the sixth and seventh transmembrane domains of PS abolishes γ-secretase activity. The proteolytic cleavage, which takes place in the middle of the transmembrane region of the substrate, occurs after ectodomain shedding of the target protein and is amino acid sequence independent. Experiments using transition state analogue inhibitors suggest that γ-secretase has separate sites for substrate binding and catalysis. Another unusual characteristic of the γ-secretase cleavage is the multiplicity of the cleavage sites: one in the middle of the transmembrane domain that creates the carboxy-terminus of Aβ, and another one that liberates intracellular domain of the substrate into the cytoplasm, for example NICD (notch site-3 cleavage) or APP ICD (AICD e-cleavage of APP). While the composition of the γ-secretase complex is still under intense investigation, three additional proteins, nicastrin, aph-1 and pen-2, have been identified so far as cofactors in the γ-secretase multicomponent complex5. Coexpression of these components with presenilin appears to be critical for γ-secretase activity. The first cofactor of the γ-secretase complex to be identified was nicastrin (NCT), a type I membrane glycoprotein comprising 709 amino acid residues in humans. Ablation of NCT in Drosophila resulted in embryonic lethality, and knockdown of NCT homolog aph-2 in C. elegans resulted in hypoplasia of the anterior pharynx. A point mutation in aph-1 gene identified in C. elegans caused an abnormality in anterior pharynx development, similar to that seen in aph-2 mutants. Disruption of the GXXXG motif within the fourth transmembrane domain of APH-1 caused by the mutation leads to failure in assembly and activation of the γ-secretase complex. Two homologues of aph-1 genes were identified in human aph-1a and aph-1b, both encoding membrane proteins predicted to span the membrane seven times. The identification of presenilin enhancer genes pen-1 and pen-2 revealed that pen-1 is identical to aph-1, while pen-2 encodes a membrane protein that spans the membrane twice. Experiments using RNAi established that suppression of the Drosophila homologues of nicastrin, APH-1 or PEN-2 result in the loss of Psn fragments and of γ-secretase activity, as indicated by lack of Aβ production. Based on systematic examination of downregulation of the different components of the γ-secretase using combinations of RNAi, it is thought that APH-1 and NCT team up to stabilize PS holoprotein, while PEN-2 induces PS endoproteolysis, eliciting maturation of the active form of the γ-secretase complex. Reconstitution of γ-secretase activity in yeast by over-coexpression of human PS1, NCT, APH-1a and PEN-2 in yeast, components which otherwise are not endogenously expressed in yeast, strongly suggests that these four cofactors are sufficient to the generation of γ-secretase activity. In contrast to other known proteases, γ-secretase is composed of a high molecular weight multicomponent complex. However, the stoichiometry of the γ-secretase is still under an intensive investigation. It is yet to be determined what other components are associated with the γ-secretase. γ-secretase regulates the second step in a two-step proteolytic cleavage event of membrane proteins known as regulated intramembrane proteolysis (RIP). This process starts with an ectodomain shedding resulting in the proteolytic processing of membrane proteins to their soluble counterparts. Ectodomain shedding is carried out by members of the ADAM family and matrix metalloproteases, and to a lesser extent by BACE1 and BACE2 (see below). Following shedding, the resulting membrane-bound CTF undergoes a second cleavage event within its transmembrane domain, called intramembrane proteolysis that is catalyzed by the γ-secretase complex.
β-Secretase
A decade ago, the enzymatic activity termed β-secretase that cleaves APP at the N’ of the Aβ region was identified as the aspartyl protease β-site APP cleaving enzyme I (BACE1; also known as memapsin and Asp2) (for review see6). A 501 amino acid type I membrane protein, BACE1 resides on the cell surface and in intracellular compartments of the secretory pathway, with its active site within the lumen of these compartments. BACE1 is predominantly expressed in neural tissues with the highest activity level exhibited in neurons. Genetic ablation of BACE1 provided an explicit evidence that BACE1 is the principal neuronal protease required for cleavage of APP at the +1 and +11 site of Aβ and the main β-secretase in the brain. BACE1 cleaves APP in its extracellular domain at a site close to the membrane surface, the Asp+1 residue, generating a secreted APP ectodomain, soluble APPβ (sAPPβ), and a membrane-bound APP C-terminal fragment of 99 amino acid residues (C99). This highly sequencespecific cleavage is a prerequisite for Aβ formation and is followed by endoproteolysis within the transmembrane domain of C99 by γ
Related posts:
 The Care Home Experience Alisoun Milne
The Care Home Experience Alisoun Milne
 The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
 The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Depression After Stroke Peter Knapp and Allan House
Sleep and Ageing: Disorders and Management Helen Chiu and Joshua Tsoh
The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Depression After Stroke Peter Knapp and Allan House
Sleep and Ageing: Disorders and Management Helen Chiu and Joshua Tsoh
 Training Requirements for Old Age Psychiatrists in the UK Susan Mary Benbow and Aparna Prasanna
Training Requirements for Old Age Psychiatrists in the UK Susan Mary Benbow and Aparna Prasanna
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


