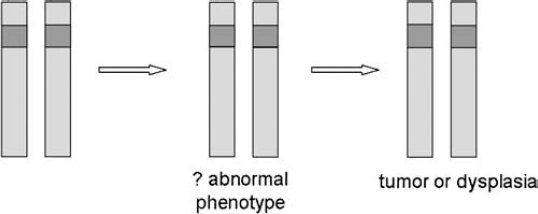
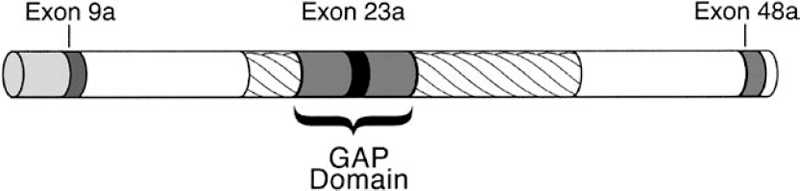
3 The neurofibromatoses are genetic disorders. NF1 and NF2 are each caused by a mutation in a known specific gene. The quest to understand how these disorders originate and progress (their pathogenesis) received a significant boost when researchers identified the causative genes. The leading theories about the pathogenesis of NF1 and NF2 are discussed in this chapter. Because the search for the biological cause of schwannomatosis was still underway when this book went to press, less is known about its pathogenesis (see Chapter 12). In 1990, two groups of scientists working separately located the NF1 gene on chromosome 17 and characterized its protein product, neurofibromin.1–3 In 1993, another two teams working separately identified the NF2 gene on chromosome 22; one named its protein “merlin”4 and the other “schwannomin.”5 Once the genes were identified, work could begin on better understanding how mutations lead to tumor formation and other manifestations. The search for answers, however, has been daunting. There is probably no single answer to the question, What causes NF1 and NF2? Just as these disorders cause various types of manifestations, so too there appear to be multiple molecular mechanisms at work. When trying to understand the pathogenesis of a disorder, scientists may combine two techniques that approach the question from different directions. The traditional phenotypical approaches analyze the physical manifestations of the disorders, such as what cells are involved and how they function, and then work backward to determine what gene might cause these abnormalities. The genotypical approach tries to understand how genes function normally and how mutations lead to the physical manifestations of disease. Many advances in knowledge about neurofibromatosis have occurred as a result of these scientific methods, but much more remains to be learned. Both the NF1 and NF2 genes are large, complex, and prone to mutation. The NF1 gene has one of the highest mutation rates of all genes.6 Even so, this is an exciting time in the field of neurofibromatosis. As research yields additional insights into the pathogenesis of NF1, NF2, and schwannomatosis, diagnosis and treatments will improve. When the NF1 and NF2 genes were first identified, both were classified as tumor suppressors. Although such genes are probably best known for their ability to prevent tumors from forming, their primary role in the body is to regulate cell growth and division. In the years since the NF1 and NF2 genes were discovered, scientists have learned more about not only how they regulate cell growth, but also the additional functions they perform in the body. Cells divide and proliferate by following an orderly and highly regulated process known as the cell cycle. The process is initiated by mitogenic signals (from the word mitosis, or division), which function a bit like on-and-off switches. When it receives such a signal, the cell initiates the cycle and copies its chromosomes, chemical structures that contain genes. After repairing any genetic errors, the cell then divides into two identical cells. When this process breaks down, cells divide out of control, crowding out other cells and forming tumors. Oncogenes are normal cellular genes that when mutated can initiate this type of abnormal growth. These genes, which help cause a variety of cancers, act in a dominant fashion. Just one mutation, or genetic “hit,” is enough to initiate the growth of a cancerous tumor. People with neurofibromatosis already have one malfunctioning gene at birth. Yet it may take years for tumors to develop, and these only rarely progress to malignancy. Figure 3–1 According to the two-hit hypothesis, one functioning copy of a tumor suppressor gene is sufficient to regulate cell proliferation, even if the other copy is defective. Only when both copies of a tumor suppressor gene become inactivated do tumors form. (From Korf BR. Human Genetics: A Problem-Based Approach. Oxford: Blackwell Science; 2000:280, with permission.) Scientists decided that the neurofibromatosis genes, therefore, seemed to conform to Alfred Knudson’s “two-hit theory” about tumor suppressors (Fig. 3–1). Knudson developed this hypothesis when describing the disease process that leads to retinoblastoma, a rare childhood cancer affecting the retina of the eye.7,8 Retinoblastoma does not occur in neurofibromatosis; the two disorders are caused by different genes. Even so, the gene responsible for retinoblastoma (the Rb gene) formed the paradigm of the tumor suppressor model, which applies to both the NFl and NF2 genes. Although both the NFl and NF2 genes function as tumor suppressors, they may inhibit cellular proliferation through different mechanisms. The NF1 gene normally suppresses cell growth directly by regulating the response to mitogenic signals. The NF2 gene works less directly, by regulating the function of signals sent between cells, and from cells to the surrounding matrix, which also inhibits or encourages cell proliferation. Both genes may also be modified or enhanced by hormones and the interaction with other genes, although this is an area deserving a great deal of rigorous study. NF1 and NF2 are both autosomal dominant disorders. Understanding what this means, and how these disorders develop, requires a basic knowledge of medical genetics. A brief introduction for the layperson is provided below. For information about why there is a 50/50 chance of passing NF1 and NF2 on to offspring, see Chapter 4. Each cell in the body contains 23 pairs of chromosomes. One member of a pair of chromosomes is inherited from the mother and the other from the father. Chromosomes 1 through 22 are known as autosomal chromosomes, and are the same in males and females. NF1 and NF2 are autosomal disorders because their genes are located on one of the autosomes. Chromosomes in the 23rd pair are known as the sex chromosomes (XX for females; XY for males). Chromosomes are located in the cell’s nucleus, which functions much like a command center that sends instructions to the rest of the cell. When genes are activated or expressed, they produce proteins that determine everything from physical characteristics, such as hair color, to less obvious traits, such as susceptibility to disease. Genes do this by providing molecular blueprints for proteins that perform various tasks. The blueprint is in the form of a unique sequence of DNA, the basic chemical building blocks of heredity (Fig. 3–2). Some genes are dominant and some are recessive—a concept first established by Gregor Mendel, the 19th century monk whose experiments with plants led to his landmark discovery of patterns of heredity. (Mendel wondered why a tall plant cross-bred with a small plant did not produce offspring of medium height. That led to the insight that certain inherited traits were dominant. The way those traits are passed on through genes would not be discovered until the next century.) Although each person has two copies of every gene, it takes two recessive genes to produce a trait, but just one dominant gene. Both NF1 and NF2 are caused by a change in a dominant gene. Whenever a cell divides, it first makes a complete copy of its DNA to pass on to the daughter cell (Fig. 3–3). Mutations can occur at any time during cell division, due to a simple copying error. Or an outside mutagen (something that can cause a mutation) such as a toxin might injure a cell’s DNA. However it happens, the mutation then gets passed on the next time the cell divides. When a DNA sequence changes because of a mutation, the genetic code gets scrambled. As a result, part of the instructions for the protein may be missing, or they may be arranged out of order. This causes some abnormality in the gene product. A truncated protein, for instance, does not contain all the necessary “ingredients” and functions poorly. At other times the protein cannot be made at all, or contains the wrong amino acid. Researchers working on the Human Genome Project estimate that people have a total of 30,000 to 40,000 genes.9 Because people inherit two copies of each gene, this means the nucleus of each cell contains about as many genes as there are words in this book. Both NF1 and NF2 develop from a mutation in just one gene (or, to use an analogy, an error in just one word in this book), which may then be exacerbated by mutations in additional “modifying” genes. The original error might be as small as the substitution of one chemical base for another in the genetic code (similar to a mistyped letter in one of the words you are reading), or as large as the deletion of the entire gene (similar to a single missing word). There are thousands of proteins in the body, which interact with one another to regulate basic cellular processes. When a genetic mutation causes one protein to malfunction, it can set off a domino-like chain reaction that affects other genes and proteins as well. Eventually this may cause manifestations of a disorder, such as tumor formation in neurofibromatosis. To determine the multiple biochemical steps between a single genetic error and the multiple manifestations of neurofibromatosis, scientists study signaling pathways—the sequence of individual genes and proteins (the dominos, if you will) that are activated or deactivated by the NF1 and NF2 genes. To do this, scientists sometimes study actual tumor tissue taken from people with NF1 or NF2, which is then examined for telltale signs of genetic mutation. Another method is to conduct studies of living cells grown in the laboratory from donated samples (the in vitro approach). Or researchers may devise experiments in vivo, in living models of the disease, such as mice or fruit flies. Generally scientists employ two techniques to better understand a gene’s function. One method is to overexpress it so that it causes more dramatic, and thus more easily observed, effects than if it were expressed normally. Another approach is to create knockout models, animals or lower organisms that are genetically engineered to lack function of one or both copies of a gene. This enables researchers to determine what happens when one or both copies of the gene are absent. Both methods provide clues about the gene’s normal function and what happens when it is mutated. The NF1 gene (Fig. 3–4) consists of ∼335,000 chemical bases,10 making it one of the largest genes in the human body. Three of the gene’s coding regions, known as exons, are alternatively spliced,11–13 meaning that sometimes they are spliced in or out of the final translated sequence. This creates a slightly different protein or isoform. This is a fairly typical feature of genes and probably enables them to take on different functions in various areas of the body. The NF1 gene also contains three “nested” but separate genes—known as EVI2A, EVI2B, and OMGP—that are embedded into one of its noncoding introns.14(p120) The function of these embedded genes is not known. Initially scientists speculated that they might have some effect on the manifestations of NF1 and might even help explain why manifestations can vary so greatly in people with the disorder. However, research in this area has been inconclusive so far. Many different types of mutations, probably more than 500 at this point, have been identified in the NF1 gene. The exact mutation usually differs from one person to the next, and only in a small number of cases have identical mutations been identified. Categories of mutations identified to date are listed in Table 3–1. Most of the mutations result in gross truncation (a shorter-than-normal version) of neurofibromin.15 Whatever the type of NF1 mutation, neurofibromin is either defective or available in such small quantities that it can’t do its job correctly. Little is known at this point about what factors activate NF1 gene expression,16(p601) but its protein product neurofibromin is expressed in several different cells throughout the body. The strongest expression is in cells of the nervous system,16(p593) including neurons and glial cells such as Schwann cells, oligodendrocytes, and astrocytes17 (see Chapter 2). Neurofibromin is also expressed in cells derived from the neural crest, a structure that is created early in embryonic development and eventually gives rise to pigment cells in the skin, bone, and some components of the peripheral nervous system. Melanocytes, for instance, produce skin pigmentation. Many nervous system and neural crest cells, of course, are also those that develop abnormalities in people with NF1.
The Pathogenesis of Neurofibromatosis 1 and Neurofibromatosis 2
♦ A Search for Answers
♦ Two Genes with Similar Functions
♦ Basic Genetics
What Genes Do
How Genes Mutate
From Genetic Error to Disorder
♦ The Pathogenesis of Neurofibromatosis 1
NF1 Gene Structure and Mutations
NF1 Gene Expression
| Mutation | Notes |
| Small deletion | Removal of a small number of DNA bases, usually leading to failure of protein production |
| Premature “stop” mutation | Changing the genetic instructions to insert an amino acid to a sequence that causes production of the protein to stop |
| Deletion of multiple exons | Can result in either shortening of the protein or complete failure of production |
| Amino acid substitution | May alter the structure or function of the protein |
| Small insertion | Has similar impact as small deletion |
| Mutation of an intron (noncoding section of a gene) | Interferes with the splicing process, resulting in an abnormal protein, or no protein produced at all |
| Deletion of entire gene | Complete gene deletion results in no protein product from that gene copy |
| Chromosome abnormality | A rearrangement of the structure of a chromosome can disrupt a gene, such as NF1 |
| Alteration of the 3′ untranslated region | Unclear if changes that follow the coding sequence of the NF1 gene are really mutations or incidental changes |
| Large insertion | Has similar impact as large deletions |
Impact on the Cell Cycle
The two prevailing theories about the pathogenesis of NF1 have something in common: that loss of the gene’s protein product somehow disrupts the normal sequence of signals responsible for controlling cell division. To appreciate how even subtle errors in molecular signaling pathways can wreak such havoc, it is important to understand the basics of the cell cycle, which initiates and controls cell division.
The cell cycle is an orderly four-step process that ends when the cell divides into two daughter cells. The cycle is regulated by a network of signaling molecules that respond to information both within and outside the cell. To use a familiar analogy, these signaling molecules are the biological versions of air traffic controllers. Some molecules monitor the status of a cell (similar to keeping an eye on a single plane); others monitor the environment (the sky or the runway). A series of signals are sent for the cell to divide (or the plane to land) only when the cell is ready and the conditions right (landing gear down, and runway clear). Otherwise, the cell will receive another set of signals that act as brakes that stop the cycle at various checkpoints. This may occur if something goes wrong during the process, or if it is simply not yet time to divide. These controls on the cycle ensure that cells wait their turn to divide and do not crowd out neighbors. Neurofibromin appears to act as a molecular brake, which is why its loss enables the cell to proliferate out of control.
NF1 Gene Signaling Pathways
Researchers are still trying to determine all of neurofibromin’s functions. Part of the challenge is the sheer size of the protein. Another is the complexity and interdependence of cells in the nervous system, which makes it difficult at times to determine which manifestations of NF1 are caused by loss of neurofibromin and which are caused by other factors. A third challenge is developing models in animals and lower organisms that mimic manifestations of NF1. Mice genetically engineered to have one functioning and one defective NF1 gene, to mimic the genetics of the disorder, do not develop tumors typical of NF1. Experiments in chimeric mice, bred to contain a defective NF1 gene as well as other features of the disorder, are helping researchers to create more accurate animal models of the disorder. Also useful are “conditional knockout” mice, in which both copies of the NF1 gene can be switched off by the investigator.
In spite of these challenges, researchers have gained much insight into neurofibromin’s functions by tracing the series of biochemical events that are triggered when the NF1 gene is expressed. This has helped researchers to learn what happens when the gene is mutated.
The Ras-GAP Pathway
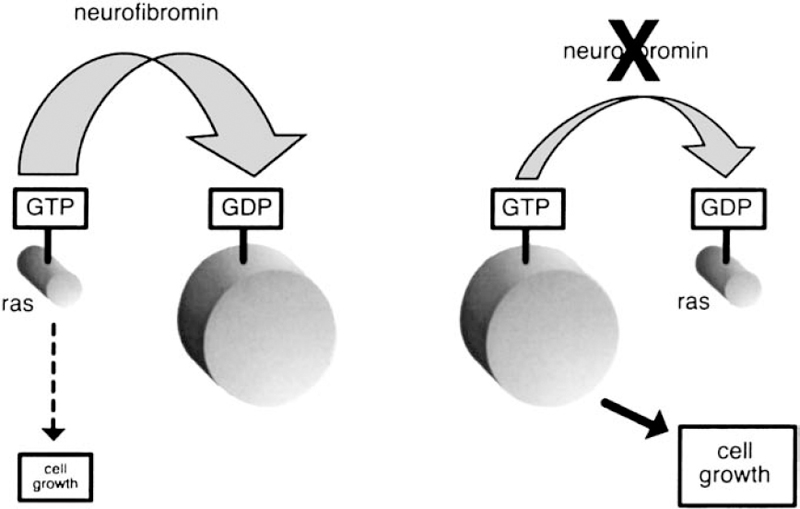
The prevailing theory about how neurofibromin functions in the body is that it behaves like a guanosine triphosphatase (GTPase)-activating protein (GAP). Members of the GAP family regulate other proteins, including one known as Ras, which is the specific target for neurofibromin GAP activity. There are more than 50 members in the Ras family,14(p126) and these proteins are best known for their role in initiating cell proliferation.18(p14) Several studies suggest that Ras also has a role in cell differentiation—the process by which cells develop specialized functions.19,20 This means that neurofibromin interaction with Ras may have an impact not only on tumor growth but also on basic development.16(p594)
Researchers first suspected that neurofibromin might regulate Ras when they noticed that a small segment of the NF1 gene resembles the sequence of genes in the GAP family.21(p748) Several functional studies have shown that neurofibromin does indeed regulate Ras.16(p593) Neurofibromin serves to act as a brake on cell proliferation, whereas Ras functions more as a gas pedal. The two are constantly in play to keep cell growth and proliferation balanced and under control. But when the NF1 gene is mutated, there is not enough neurofibromin to counter the actions of Ras. The result is out-of-control cell growth and tumor formation (Fig. 3–5).
Related posts:
 Cognitive Function and Learning Disabilities in Neurofibromatosis 1
Cognitive Function and Learning Disabilities in Neurofibromatosis 1
 Psychosocial Impact of Neurofibromatosis
Psychosocial Impact of Neurofibromatosis
 Genetics and Genetic Counseling in Neurofibromatosis 1 and Neurofibromatosis 2
Genetics and Genetic Counseling in Neurofibromatosis 1 and Neurofibromatosis 2
 Diagnosis and Overall Management of Neurofibromatosis 2
Diagnosis and Overall Management of Neurofibromatosis 2
 The Many Faces of Neurofibromatosis
The Many Faces of Neurofibromatosis
 Other Features of Neurofibromatosis 1
Other Features of Neurofibromatosis 1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


